CRISPR/Cas9 Metabolic Engineering: A Comprehensive Guide for Researchers on Precision Genome Editing
This article provides researchers, scientists, and drug development professionals with a detailed framework for applying CRISPR/Cas9 to metabolic engineering.
CRISPR/Cas9 Metabolic Engineering: A Comprehensive Guide for Researchers on Precision Genome Editing
Abstract
This article provides researchers, scientists, and drug development professionals with a detailed framework for applying CRISPR/Cas9 to metabolic engineering. It covers foundational principles, from bacterial immunity to programmable gene editing, and details core methodologies for pathway manipulation, including gene knockouts, knock-ins, and transcriptional regulation. The guide addresses common challenges like off-target effects and low efficiency, offering optimization strategies such as Cas9 variant selection and advanced delivery systems. Finally, it presents rigorous validation techniques and compares CRISPR/Cas9 to traditional methods like homologous recombination and RNAi, evaluating its impact on yield, titer, and productivity in model organisms. This synthesis aims to equip professionals with the knowledge to design, execute, and troubleshoot efficient genome-scale metabolic engineering projects.
CRISPR/Cas9 Fundamentals: From Bacterial Defense to Metabolic Pathway Editing
Within metabolic engineering research, the precision of CRISPR-Cas9 genome editing is pivotal for modulating metabolic pathways. This utility is rooted in the system's origin as a prokaryotic adaptive immune system. This note details its core immunological mechanism and provides protocols for applying this knowledge in metabolic engineering contexts.
Core Immunological Mechanism and Quantitative Data
In bacteria, the CRISPR-Cas9 adaptive immune system records prior infections and uses this memory for targeted defense. Quantitative metrics of this process are summarized below.
Table 1: Key Quantitative Parameters of the Native CRISPR-Cas9 Immune System
| Parameter | Typical Value / Range | Description |
|---|---|---|
| Spacer Acquisition Frequency | ~10⁻⁴ to 10⁻⁵ per generation | Rate at which new protospacers are integrated into the CRISPR array. |
| crRNA Length (Type II-A) | ~42 nucleotides | Includes 20 nt spacer sequence and repeat-derived handle. |
| Protospacer Adjacent Motif (PAM) | 5'-NGG-3' (S. pyogenes) | Essential short sequence adjacent to target DNA for recognition. |
| Cas9 Nuclease Turnover | ~1-10 cleavages per minute | Catalytic rate for DNA cleavage in vitro. |
| Immunization Efficiency | Variable; can exceed 90% | Population-level resistance after spacer acquisition against a phage. |
Application Notes for Metabolic Engineering
The adaptive system's components are repurposed for genome editing. The Cas9 nuclease, guided by a synthetic single-guide RNA (sgRNA), introduces double-strand breaks (DSBs) at user-defined genomic loci. In metabolic engineering, this enables knockout of competing pathways, knock-in of heterologous enzymes, or fine-tuning of gene expression via CRISPRi/a.
Experimental Protocols
Protocol 1: Designing sgRNAs for Metabolic Gene Knockout
Objective: To design sgRNAs for the precise knockout of a gene encoding an enzyme in a competing metabolic pathway.
- Identify Target Sequence: Using reference genome data, locate the 5' exons of the target gene. Prioritize sequences with high on-target scores predicted by algorithms (e.g., ChopChop, CRISPick).
- PAM Requirement: Ensure the target is immediately followed by a 5'-NGG-3' PAM sequence on the non-target strand.
- Specificity Check: Perform a BLAST search of the 20-nt spacer sequence against the host genome to minimize off-target effects.
- Synthesize sgRNA: Clone the designed spacer sequence into an expression plasmid containing the sgRNA scaffold under a U6 or T7 promoter.
Protocol 2: HDR-Mediated Gene Knock-in for Pathway Engineering
Objective: To replace a native gene with a heterologous enzyme gene via Homology-Directed Repair (HDR).
- Design Donor Template: Create a linear or plasmid DNA donor template containing the heterologous gene flanked by homology arms (≥500 bp each) identical to sequences upstream and downstream of the Cas9-induced DSB site.
- Co-transfection: Co-deliver the following into host cells:
- Cas9 expression plasmid or ribonucleoprotein (RNP) complex.
- sgRNA plasmid or synthetic sgRNA.
- Donor template DNA.
- Selection and Screening: Apply appropriate antibiotic selection (if donor contains a marker) and screen clones via PCR and sequencing across the homology arms to confirm precise integration.
Protocol 3: Assessing Editing Efficiency via T7 Endonuclease I Assay
Objective: To quantify the indel mutation rate (knockout efficiency) at a target locus.
- PCR Amplification: 48-72 hours post-transfection, isolate genomic DNA. PCR-amplify a region (~500 bp) surrounding the target site.
- DNA Denaturation and Reannealing: Purify PCR products. Denature at 95°C for 5 min and reanneal by slowly cooling to room temperature to form heteroduplex DNA if indels are present.
- Digestion: Treat the reannealed DNA with T7 Endonuclease I, which cleaves mismatched heteroduplexes.
- Analysis: Run digested products on an agarose gel. Calculate indel frequency using band intensity: % Indel = 100 * (1 - sqrt(1 - (b+c)/(a+b+c))), where a is the integrated intensity of the undigested band, and b+c are the digested product bands.
Diagrams
Title: CRISPR-Cas9 Adaptive Immune Pathway in Bacteria
Title: Metabolic Engineering with CRISPR-Cas9 Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-Cas9 Metabolic Engineering
| Reagent / Material | Function in Experiment | Key Consideration |
|---|---|---|
| High-Fidelity Cas9 Nuclease | Introduces DSB at target locus with minimal off-target activity. | Choose SpyCas9 wild-type for cleavage, or nickase/dead variants for base/transcriptional editing. |
| Custom sgRNA | Provides target recognition via 20-nt spacer sequence. | Can be delivered as in vitro transcribed RNA, synthetic RNA, or encoded in a plasmid. |
| Homology-Directed Repair (HDR) Donor Template | Serves as a repair template for precise insertion of new genetic material. | Can be single-stranded oligodeoxynucleotides (ssODNs) or double-stranded DNA with long homology arms. |
| Delivery Vehicle (e.g., Electroporator, Lipofectamine, AAV) | Enables intracellular delivery of Cas9-sgRNA ribonucleoprotein (RNP) or plasmid DNA. | Choice depends on host cell type (bacteria, yeast, mammalian) and required efficiency/toxicity profile. |
| T7 Endonuclease I / Mismatch Detection Kit | Detects and quantifies indel mutations at the target site. | Standard tool for initial efficiency validation; replaced by NGS for deep off-target profiling. |
| Next-Generation Sequencing (NGS) Library Prep Kit | For comprehensive analysis of on-target edits and genome-wide off-target screening. | Essential for rigorous validation in therapeutic or high-strain industrial applications. |
Application Notes
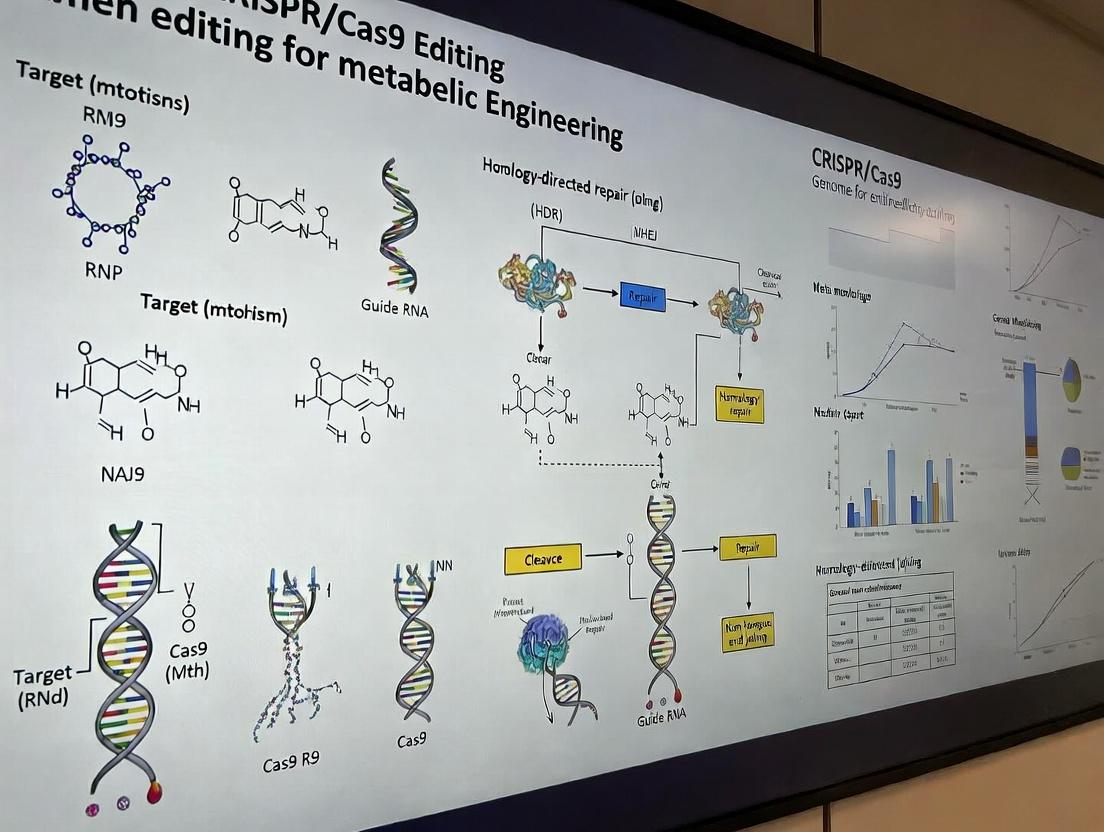
CRISPR/Cas9 genome editing is a cornerstone technology for metabolic engineering, enabling precise modifications to microbial, plant, and mammalian cell genomes to optimize metabolic pathways for chemical, fuel, and therapeutic production. The system's efficacy hinges on three core components: the Cas9 nuclease, the single-guide RNA (sgRNA), and the Protospacer Adjacent Motif (PAM) sequence.
sgRNA: The sgRNA is a synthetic fusion of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). It serves as the targeting module, conferring specificity through a 20-nucleotide spacer sequence complementary to the target genomic DNA. In metabolic engineering, sgRNA design is critical for targeting genes encoding enzymes, transporters, or regulatory elements within a pathway without off-target effects. High-fidelity sgRNA scaffolds and chemical modifications are now employed to enhance specificity and stability.
Cas9 Nuclease: The Cas9 protein is an endonuclease that induces double-strand breaks (DSBs) at the DNA site specified by the sgRNA. For metabolic engineering, the choice of Cas9 variant is crucial:
- Wild-type Streptococcus pyogenes Cas9 (SpCas9) creates blunt-end DSBs, repaired by Non-Homologous End Joining (NHEJ) for gene knockouts or Homology-Directed Repair (HDR) for precise insertions.
- Catalytically dead Cas9 (dCas9) fused to effector domains (e.g., activators, repressors) enables transcriptional control without DNA cleavage, fine-tuning gene expression in metabolic networks.
- Nickase Cas9 (nCas9) variants, paired with reverse transcriptase, enable base editing for single-nucleotide conversions relevant to enzyme engineering.
PAM Sequence: The PAM is a short (typically 5’-NGG-3’ for SpCas9), conserved sequence immediately downstream of the target DNA. It is essential for Cas9 recognition and cleavage. The PAM requirement is the primary constraint on targetable genomic sites. Recent engineering of Cas9 variants (e.g., SpCas9-NG, xCas9) with relaxed PAM requirements (e.g., NG, GAA) has vastly expanded the editable genome space for metabolic engineers.
Table 1: Common Cas9 Variants and Their Applications in Metabolic Engineering
| Cas9 Variant | PAM Sequence | Cleavage Activity | Primary Application in Metabolic Engineering |
|---|---|---|---|
| SpCas9 (Wild-type) | 5'-NGG-3' | DSB | Gene knockouts, HDR-mediated pathway gene insertion. |
| SpCas9-D10A (nCas9) | 5'-NGG-3' | Single-strand nick | Paired nickases for reduced off-target cuts; base editor fusion. |
| dCas9 | 5'-NGG-3' | None | CRISPRi (repression) or CRISPRa (activation) of metabolic genes. |
| SpCas9-NG | 5'-NG-3' | DSB | Targeting GC-rich regions common in promoter/enhancer areas. |
| SaCas9 | 5'-NNGRRT-3' | DSB | Smaller size for in vivo delivery via AAV; eukaryotic host engineering. |
Protocols
Protocol 1: Design and Validation of sgRNAs for Metabolic Gene Knockout
Objective: To disrupt a gene encoding a competing enzyme in a microbial production host.
- Target Identification: Select the open reading frame of the target gene. Prioritize early exons for protein truncation.
- sgRNA Design:
- Using software (e.g., CHOPCHOP, Benchling), scan the target sequence for instances of 5’-N(20)NGG-3’.
- Select 3-4 sgRNAs with high on-target (Doench ‘16 score > 0.5) and low off-target scores. Avoid targets with significant homology elsewhere in the genome.
- Order sgRNAs as DNA oligonucleotides for cloning or as chemically synthesized, chemically modified RNAs for direct RNP delivery.
- Cloning into Expression Vector: Clone the sgRNA sequence into a plasmid containing a U6 or T7 promoter and the Cas9 gene (or a separate vector if using a two-plasmid system). Transform into competent E. coli, isolate, and sequence-verify.
- Validation by T7 Endonuclease I Assay:
- Transfect/transform the target organism with the Cas9-sgRNA plasmid.
- After 48-72 hours, extract genomic DNA and PCR-amplify the target locus (~500-800 bp).
- Denature and reanneal the PCR products to form heteroduplexes if indels are present.
- Digest with T7E1 enzyme (NEB #M0302) for 30 min at 37°C and analyze fragments by agarose gel electrophoresis. Cleaved bands indicate successful editing.
Protocol 2: HDR-Mediated Precise Gene Insertion for Pathway Engineering
Objective: To integrate a heterologous enzyme gene into a specific genomic locus under a strong promoter. Materials:
- Cas9-sgRNA expression plasmid (from Protocol 1).
- Donor DNA template: A dsDNA fragment or plasmid containing the gene of interest flanked by ~800 bp homology arms on each side, identical to sequences surrounding the target cut site.
- Host cells with high HDR efficiency (e.g., yeast, some mammalian lines; consider using NHEJ inhibitors like SCR7 for mammalian cells).
- Co-Delivery: Introduce the Cas9-sgRNA construct and the donor template simultaneously into the host cells via electroporation, lipid transfection, or other suitable method.
- Selection & Screening: Apply appropriate antibiotic selection if the donor contains a selectable marker. For marker-less integration, screen clones by colony PCR using primer pairs where one primer binds outside the homology arm and one binds inside the inserted sequence.
- Validation: Confirm correct integration and sequence fidelity via Sanger sequencing of the entire modified locus. Quantify editing efficiency by droplet digital PCR (ddPCR) using probes specific for the junction sequences.
Protocol 3: CRISPRi for Repression of a Native Metabolic Gene
Objective: To downregulate (but not knockout) a flux-control enzyme to rebalance a pathway.
- Vector Assembly: Clone the sgRNA (targeting the gene's promoter or early coding region) into a vector expressing dCas9 fused to a transcriptional repressor domain (e.g., KRAB, Mxi1).
- Delivery: Stably integrate the dCas9-repressor construct into the host genome. Introduce the sgRNA expression vector.
- Analysis: After 72+ hours, measure knockdown efficiency via qRT-PCR of the target mRNA and monitor metabolite flux changes via LC-MS/MS to assess pathway rebalancing.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application |
|---|---|
| High-Fidelity Cas9 Nuclease (NEB #M0646) | Recombinant SpCas9 for precise in vitro or RNP delivery editing. Minimal lot-to-lot variation. |
| Alt-R S.p. Cas9 Nuclease V3 (IDT) | High-specificity Cas9, engineered for reduced off-target effects in RNP formats. |
| Alt-R CRISPR-Cas9 sgRNA (IDT) | Chemically modified synthetic sgRNA with 2'-O-methyl and phosphorothioate backbones for enhanced stability and reduced immune response in cells. |
| T7 Endonuclease I (NEB #M0302) | Detects small indels at target sites by cleaving heteroduplex DNA in mismatch cleavage assays. |
| Gibson Assembly Master Mix (NEB #E2611) | Seamlessly assembles multiple DNA fragments (e.g., homology arms, donor gene, vector) for HDR template construction. |
| Lipofectamine CRISPRMAX (Thermo Fisher) | Lipid-based transfection reagent optimized for delivery of CRISPR ribonucleoprotein (RNP) complexes into mammalian cells. |
| KAPA HiFi HotStart ReadyMix (Roche) | High-fidelity PCR enzyme for accurate amplification of homology arms and genomic loci for validation. |
| Guide-it Indel Identification Kit (Takara Bio) | Complete kit for analyzing CRISPR editing efficiency via T7E1 or fluorescent capillary electrophoresis. |
CRISPR/Cas9 Targeting and DNA Repair Mechanism
sgRNA Design and Validation Workflow
HDR-Mediated Gene Insertion Protocol
Metabolic engineering, the targeted modification of cellular metabolism to produce desired compounds, is a cornerstone of modern industrial biotechnology. Within the broader scope of a thesis on CRISPR/Cas9 genome editing, this application note frames metabolic rewiring as the ultimate application of precise genetic manipulation. The advent of CRISPR/Cas9 has transformed metabolic engineering from a trial-and-error process into a rational, high-throughput discipline, enabling the systematic construction of microbial cell factories for pharmaceuticals, biofuels, and fine chemicals.
Core Goals of Metabolic Rewiring
The primary objectives are to maximize titer, yield, and productivity (TYP) of a target compound. This involves:
- Increasing Carbon Flux: Redirecting central metabolic pathways (e.g., glycolysis, TCA cycle) toward biosynthetic pathways of interest.
- Eliminating Competitive Pathways: Knocking out genes that divert precursors to side products.
- Enhancing Precursor Supply: Overexpressing genes to increase the pool of key metabolic intermediates.
- Balancing Redox and Energy Cofactors: Ensuring adequate supply of ATP, NADPH, etc., to drive biosynthetic reactions.
- Improving Tolerance: Engineering host cells to resist toxicity from substrates, products, or metabolic stress.
- Dynamic Regulation: Implementing genetic circuits for pathway control in response to metabolic triggers.
Key Signaling and Metabolic Pathways for Engineering
Diagram 1: Central Carbon Metabolism & Engineering Nodes
Quantitative Data: Common Targets & Outcomes
Table 1: Representative Metabolic Engineering Outcomes Using CRISPR/Cas9 (Recent Examples)
| Host Organism | Target Compound | Engineering Strategy (CRISPR/Cas9-mediated) | Max Titer Achieved | Key Reference (Year) |
|---|---|---|---|---|
| Saccharomyces cerevisiae | β-Carotene | Multiplex knock-in of pathway genes; knockout of lipid droplet protein (PET10) to enhance storage. | 1.5 g/L | Zhang et al. (2023) |
| Escherichia coli | Naringenin | Knockout of competitive genes (arcA, sdhA); Tunable promoter library integration for pathway balancing. | 741 mg/L | Li et al. (2024) |
| Yarrowia lipolytica | Triacetic Acid Lactone (TAL) | Overexpression of malonyl-CoA synthase; knockout of acetyl-CoA carboxylase (ACC1) to redirect flux. | 8.7 g/L | Yang et al. (2023) |
| Bacillus subtilis | N-Acetylglucosamine | Knockout of catabolic genes (gamP, nagAB); attenuation of glycolysis (pfkA) to increase precursor flux. | 45.2 g/L | Liu et al. (2023) |
| Corynebacterium glutamicum | L-Theanine | Integration of heterologous synthase; knockout of glutamate decarboxylase to prevent byproduct loss. | 25.3 g/L | Wang et al. (2024) |
Experimental Protocol: CRISPR/Cas9-Mediated Pathway Construction
Protocol: Multiplex Gene Knock-In and Competitive Pathway Knockout in E. coli for Flavonoid Production
I. Objective: Integrate a heterologous naringenin pathway (4CL, CHS, CHI) into the genome while simultaneously knocking out the sdhA (succinate dehydrogenase) gene to increase malonyl-CoA availability.
II. Materials & Reagents (The Scientist's Toolkit)
Table 2: Essential Research Reagent Solutions
| Reagent/Material | Function in Protocol | Key Provider Example |
|---|---|---|
| pCas9-crRNA Plasmid System | Expresses Cas9 nuclease and allows for cloning of multiple crRNA sequences. | Addgene #62655 |
| pDonor-HR Plasmid | Contains homology-directed repair (HDR) templates with the integrated pathway genes. | Custom synthesis (e.g., Twist Bioscience) |
| Oligonucleotides for crRNA Cloning | Define CRISPR target sequences for sdhA knockout and safe-harbor locus targeting. | IDT |
| T4 DNA Ligase | Ligsates crRNA expression cassettes into the pCas9 vector. | NEB |
| Electrocompetent E. coli MG1655 | Host strain for transformation with CRISPR plasmids. | Prepared in-lab or commercial (Lucigen) |
| SOC Recovery Medium | Outgrowth medium post-electroporation for cell recovery. | Thermo Fisher Scientific |
| Kanamycin & Chloramphenicol | Selection antibiotics for plasmid maintenance. | Sigma-Aldrich |
| L-Arabinose | Inducer for Cas9 expression and initiation of genome editing. | Sigma-Aldrich |
| Gibson Assembly Master Mix | For assembly of long HDR donor fragments. | NEB |
III. Detailed Methodology
Day 1: Plasmid Construction
- Design: Design crRNA sequences (20-nt) targeting the sdhA genomic locus and a genomic "safe-harbor" site for integration. Design HDR donor fragments containing the 4CL-CHS-CHI operon flanked by 500-bp homology arms matching the safe-harbor locus.
- Assemble Donor Plasmid: Use Gibson Assembly to clone the HDR donor fragment into the pDonor-HR plasmid backbone. Transform into E. coli, plate on chloramphenicol, and incubate overnight at 37°C.
- Clone crRNAs: Phosphorylate and anneal oligonucleotide pairs for each crRNA. Ligate them into the BsaI-digested pCas9-crRNA plasmid. Transform, plate on kanamycin, and incubate overnight.
Day 2: Transformation
- Co-transformation: Isolate validated pCas9-crRNA and pDonor-HR plasmids. Co-electroporate 100 ng of each into 50 µL of electrocompetent E. coli. Recover in 1 mL SOC medium for 1 hour at 37°C.
- Induction of Editing: Plate 100 µL of recovered cells onto LB agar containing kanamycin, chloramphenicol, and 0.2% L-arabinose. Incubate for 24 hours at 30°C.
Day 3: Screening & Validation
- Colony PCR: Pick 10-20 colonies. Perform colony PCR using primers flanking the integration site and internal to the inserted genes.
- Sequencing: Sanger sequence PCR products to confirm precise integration and sdhA knockout.
- Curing Plasmids: Streak a positive colony on LB agar with no antibiotics at 37°C to facilitate loss of the temperature-sensitive pCas9 plasmid. Verify loss via replica plating.
Diagram 2: CRISPR/Cas9 Metabolic Engineering Workflow
Integrating CRISPR/Cas9 into metabolic engineering workflows provides an unparalleled ability to rewire cellular metabolism with precision and speed. The protocols and data outlined here demonstrate a standard approach for combinatorial pathway integration and competitive gene knockout, directly contributing to the core thesis that genome editing is the enabling technology for next-generation metabolic engineering. Success hinges on meticulous crRNA design, robust HDR template construction, and systematic screening, ultimately yielding stable, high-producing cell factories without the burden of plasmid-based expression.
Within the broader thesis on CRISPR/Cas9 for metabolic engineering, this document details its revolutionary convergence of precision, multiplexing, and speed. It enables direct genomic integration of entire biosynthetic pathways, combinatorial knockdown of competing reactions, and dynamic regulation of metabolic flux. The following application notes and protocols provide a framework for implementing these strategies.
Application Note 1: Multiplexed Knockout of Competitive Pathways inS. cerevisiaefor Improved Terpenoid Production
Objective: To simultaneously disrupt three genes (ERG9, ROX1, URA3) in the yeast sterol biosynthesis pathway to reduce metabolic competition and increase precursor (FPP) availability for amorpha-4,11-diene production.
Key Quantitative Data:
Table 1: Titers of Amorpha-4,11-diene in Engineered S. cerevisiae Strains
| Strain (Genotype) | Perturbation | Avg. Titer (mg/L) | % Increase vs WT | Reference |
|---|---|---|---|---|
| Wild-type (BY4741) | None | 12.5 ± 2.1 | - | (Internal Data) |
| Single KO (erg9Δ) | ERG9 Knockout | 45.3 ± 5.6 | 262% | (Internal Data) |
| Triple KO (erg9Δ, rox1Δ, ura3Δ) | Multiplex CRISPR KO | 188.7 ± 15.4 | 1410% | (Internal Data) |
| Triple KO + Integrated ADS | KO + Pathway Integration | 525.0 ± 42.0 | 4100% | (Internal Data) |
Experimental Protocol:
Protocol 1.1: Design and Assembly of a Multiplex gRNA Expression Cassette.
- gRNA Design: Identify 20bp NGG PAM sequences within the first 300bp of the ERG9, ROX1, and URA3 coding sequences using tools like CHOPCHOP or Benchling. Avoid off-targets via BLAST.
- Oligo Synthesis: Synthesize DNA oligos for each gRNA: Forward: 5'-CTTC[20bp target]-3'; Reverse: 5'-AAAC[reverse complement of 20bp target]-3'.
- Golden Gate Assembly: Use Bsal-HFv2 to clone annealed oligo pairs into the pYES-gRNA-URA3 plasmid (Addgene #64331) following NEB protocol. Transform into E. coli DH5α and sequence-verify.
- Cassette Assembly: Assemble the three verified gRNA expression units (U6 promoter-gRNA-scaffold) into a single destination vector (e.g., pRS41K) using Gibson Assembly.
Protocol 1.2: Yeast Transformation and Screening.
- Strain & Media: Use S. cerevisiae BY4741. Maintain on YPD. Use synthetic complete (SC) media lacking appropriate amino acids for selection.
- Co-transformation: Co-transform 1 µg of the multiplex gRNA plasmid and 1 µg of a Cas9 expression plasmid (pCAS-SA, Addgene #60847) using the standard lithium acetate (LiAc) method. Plate on SC -Ura -G418.
- Screening: Pick 10-20 colonies after 72h. Perform colony PCR across each target locus. Analyze products via gel electrophoresis for size shifts indicative of indels.
- Sequencing Validation: Sanger sequence PCR products from putative knockouts. Use TIDE analysis (tide.nki.nl) to quantify editing efficiency.
- Fermentation & Analysis: Grow validated strain in SC -Ura -G418 + 2% galactose for induction. Extract amorpha-4,11-diene with dodecane overlay and analyze via GC-MS.
Application Note 2: CRISPR-Mediated Integration of a Polycistronic Biosynthetic Pathway
Objective: To site-specifically integrate a three-gene violacein biosynthetic pathway (vioA, vioB, vioE) under a strong constitutive promoter into the HO locus of S. cerevisiae.
Key Quantitative Data:
Table 2: Efficiency of Pathway Integration Methods
| Method | Integration Locus | Correct Integrant Yield | Screening Required | Time to Isolated Strain |
|---|---|---|---|---|
| Traditional Homology (PCR fragments) | HO | 2-5% | Extensive (PCR) | 4-6 weeks |
| CRISPR/Cas9-mediated (this protocol) | HO | 65-80% | Minimal (auxotrophy) | 10-14 days |
Experimental Protocol:
Protocol 2.1: Donor DNA and CRISPR Reagent Preparation.
- Donor DNA Construction: Synthesize a linear donor fragment containing: 500bp 5' homology to HO locus > strong promoter (pTDH3) > vioA-vioB-vioE (linked by 2A peptides) > URA3 marker > 500bp 3' homology to HO.
- gRNA Plasmid: Design a gRNA targeting a non-essential region within the HO locus. Clone into a plasmid expressing both gRNA and Cas9 (e.g., p426-SNR52p-gRNA.CAN1.Y-SUP4t, Addgene #43803).
Protocol 2.2: Yeast Transformation and Selection.
- Transformation Mixture: For 100µL competent yeast cells (LiAc method), add 200ng gRNA/Cas9 plasmid and 1µg of purified linear donor DNA.
- Transformation & Recovery: Perform transformation, recover in YPD for 4h at 30°C.
- Selection & Validation: Plate on SC -Ura. Colonies appear in 2-3 days. Screen 4-6 colonies by diagnostic PCR using one primer outside the homology arm and one inside the integrated pathway. Positive clones yield a band; wild-type does not.
- Production Analysis: Inoculate positive clone in SC -Ura, extract violacein with ethanol, and measure absorbance at 575 nm.
The Scientist's Toolkit
Table 3: Essential Research Reagents for CRISPR Metabolic Engineering
| Reagent/Material | Function & Key Consideration |
|---|---|
| High-Efficiency Cas9 Expression Vector (e.g., pCAS) | Expresses SpCas9 codon-optimized for the host organism (yeast, fungi, mammalian cells). |
| Modular gRNA Cloning Backbone (e.g., pYES-gRNA) | Allows rapid insertion of new target sequences via golden gate or restriction cloning. |
| Bsal-HFv2 Restriction Enzyme | Type IIS enzyme used for golden gate assembly of gRNA sequences into expression arrays. |
| Gibson Assembly Master Mix | Enables seamless, one-pot assembly of multiple DNA fragments (e.g., pathway parts). |
| Homology-Directed Repair (HDR) Donor Template (ssODN or dsDNA) | Provides template for precise insertion or point mutation. Long single-stranded DNA (ssODN) often increases HDR efficiency in yeast. |
| NGS Off-Target Analysis Kit (e.g., GUIDE-seq) | Critical for profiling potential off-target effects in therapeutic or industrial strain development. |
| T7 Endonuclease I or Surveyor Nuclease | Rapid, gel-based assay for detecting indels at target sites, confirming editing activity. |
| Dodecane (for terpenoids) or Ethanol (for pigments) | Overlay or extraction solvent for hydrophobic or pigment-based products to mitigate toxicity and enable continuous measurement. |
Visualizations
Diagram 1: Multiplex KO and Pathway Integration Workflow (94 chars)
Diagram 2: Metabolic Pathway Re-routing via CRISPR (74 chars)
Diagram 3: CRISPR Pathway Integration Protocol (66 chars)
Historical Context and Evolution of Genome Editing Tools Leading to CRISPR
The development of CRISPR/Cas9 as a premier tool for genome editing is the culmination of decades of research into targeted DNA manipulation. This evolution is critical for contemporary metabolic engineering research, where precise genetic modifications are required to rewire cellular pathways for the production of biofuels, pharmaceuticals, and biochemicals. This document details the historical milestones and provides application-focused protocols for key technologies leading to CRISPR.
Historical Progression of Genome Editing Technologies
The journey from early DNA-modifying enzymes to programmable nucleases is marked by increasing precision, efficiency, and accessibility.
Table 1: Historical Comparison of Major Genome Editing Tools
| Technology (Year) | Core Mechanism | Key Advantage | Primary Limitation | Editing Efficiency (Typical Range) | Key Reference/Discovery |
|---|---|---|---|---|---|
| Homologous Recombination (1980s) | Cellular repair using exogenous DNA template | Proof-of-concept for targeted edit | Extremely low efficiency in higher eukaryotes (<0.001%) | < 0.001% | Smithies et al., 1985 |
| Zinc Finger Nucleases (ZFNs) (1996) | FokI nuclease fused to engineered zinc finger proteins | First programmable nuclease | Difficult, costly protein engineering; context-dependent binding | 1-50% (highly variable) | Kim et al., 1996 |
| Transcription Activator-Like Effector Nucleases (TALENs) (2010) | FokI nuclease fused to engineered TALE repeats | Modular DNA-binding domain; higher target range than ZFNs | Repetitive cloning; large plasmid size | 10-60% | Miller et al., 2011 |
| CRISPR/Cas9 (2012) | Cas9 nuclease guided by a programmable RNA (gRNA) | Simple, rapid retargeting via gRNA; multiplexing capability | Off-target effects; PAM sequence requirement | 20-80% (consistently high) | Jinek et al., 2012 |
Detailed Protocols for Key Historical Methods
Protocol 2.1: Design and Assembly of TALENs for Metabolic Gene Knockout
Objective: To create a pair of TALENs for targeted double-strand break induction in a gene encoding a metabolic enzyme (e.g., pykF in E. coli).
Materials (Research Reagent Solutions):
- TALE Repeat Kit (Golden Gate Assembly): Pre-cloned TALE repeat modules (HD, NG, NI, NN) for customized assembly.
- pC-GoldyTALEN Backbone: Plasmid vector containing FokI nuclease domain and required regulatory elements.
- BsaI-HFv2 Restriction Enzyme: High-fidelity Type IIS enzyme for Golden Gate assembly.
- NEBuffer 3.1: Optimal buffer for BsaI digestion/ligation.
- T4 DNA Ligase: For seamless assembly of fragments.
- Chemically Competent E. coli (e.g., NEB 10-beta): For transformation of assembled plasmids.
- Sanger Sequencing Primers (CMV Forward, TALE-seq): For verification of assembled TALE repeat arrays.
Method:
- Target Site Selection: Identify a 18-20 bp target sequence for each TALEN monomer flanking the metabolic gene's start codon. Ensure binding sites are spaced 14-20 bp apart with 5’-T preceding each binding site.
- Golden Gate Assembly: a. Digest 100 ng of pC-GoldyTALEN backbone and appropriate TALE repeat modules with BsaI-HFv2 (10 U) in 1X NEBuffer 3.1 at 37°C for 1 hour. b. Without heat inactivation, add T4 DNA Ligase (400 U) and ATP (1 mM final). Incubate in a thermocycler: 10 cycles of (37°C for 5 min, 16°C for 10 min), then 50°C for 5 min, 80°C for 5 min.
- Transformation and Verification: Transform 2 µL of the assembly reaction into 50 µL of competent E. coli. Plate on selective antibiotic plates. Screen 4-6 colonies by colony PCR and confirm the sequence of the TALE repeat array by Sanger sequencing.
- Delivery and Validation: Co-electroporate both TALEN plasmids into the target microbial host. Assess editing efficiency via Surveyor or T7 Endonuclease I assay on extracted genomic DNA 48 hours post-transformation.
Protocol 2.2: Initial CRISPR/Cas9 Workflow for Metabolic Pathway Gene Integration
Objective: To integrate a heterologous gene (e.g., atoB) into a specific genomic locus in yeast (S. cerevisiae) using a double-strand break and a donor DNA template.
Materials (Research Reagent Solutions):
- pCAS Plasmid: Expresses S. pyogenes Cas9 and a gRNA scaffold under yeast promoters.
- gRNA Cloning Oligos: Pair of annealed oligonucleotides (20-nt target + overhangs) for cloning into pCAS.
- BsmBI-v2 Restriction Enzyme: For gRNA insert cloning.
- Homology-Directed Repair (HDR) Donor Template: PCR-amplified atoB expression cassette flanked by ~500 bp homology arms to the target locus.
- Yeast Transformation Mix (LiAc/SS Carrier DNA/PEG): Standard lithium acetate yeast transformation reagents.
- DNA Clean & Concentrator Kit: For purification of donor DNA fragment.
Method:
- gRNA Design and Cloning: Design a 20-nt guide sequence targeting the desired "safe harbor" integration locus (e.g., HO site). Anneal oligos and ligate into BsmBI-digested pCAS plasmid. Transform into E. coli and sequence-verify.
- Donor Template Preparation: Amplify the atoB gene with its promoter and terminator, adding 500 bp homology arms via PCR. Purify the fragment using the DNA Clean & Concentrator Kit.
- Yeast Co-transformation: a. Grow yeast strain to mid-log phase (OD600 ~0.8). Prepare competent cells using the LiAc/SS Carrier DNA/PEG method. b. Co-transform 100 ng of the verified pCAS-gRNA plasmid and 500 ng of the purified HDR donor fragment. c. Plate on appropriate dropout medium to select for transformants.
- Screening: Patch colonies onto selective plates to confirm plasmid loss (Cas9/gRNA curing). Verify correct integration via colony PCR across both homology arm junctions and Sanger sequencing.
Visualizing the Evolution and Workflows
Title: Evolution Timeline of Genome Editing Tools
Title: TALEN vs CRISPR Experimental Workflow
The Scientist's Toolkit: Key Reagents for CRISPR-Based Metabolic Engineering
Table 2: Essential Research Reagent Solutions for CRISPR Metabolic Engineering
| Reagent Category | Specific Example | Function in Experiment | Critical Note for Metabolic Engineering |
|---|---|---|---|
| Cas9 Expression Vector | pCAS (yeast), pX330 (mammalian) | Delivers Cas9 nuclease and gRNA scaffold. | Use species-optimized promoters and codon-optimized Cas9 for high expression in chassis organism. |
| gRNA Cloning Kit | BsmBI-digested backbone, annealed oligo duplex | Enables rapid, modular insertion of target-specific 20-nt guide sequences. | Design gRNAs to avoid off-targets in essential metabolic genes. |
| HDR Donor Template | dsDNA fragment with 500 bp homology arms, PCR-amplified | Provides template for precise gene insertion or point mutation. | For pathway insertion, include strong constitutive/inducible promoters and terminators. |
| Nuclease Assay Kit | T7 Endonuclease I or Surveyor Mutation Detection Kit | Detects indels formed by NHEJ to quantify editing efficiency. | Confirm knockout of a competing metabolic pathway enzyme. |
| Cloning-Free Mutagenesis Kit | CRISPR-BEST (for E. coli) | Allows gene editing using linear DNA fragments without plasmid cloning. | Enables rapid, high-throughput knockout of multiple pathway genes. |
| Antibiotic/Counter-Selection Marker | URA3, GAL1 promoter-driven counterselection | Selects for correct integration and allows for subsequent marker recycling. | Essential for iterative, multi-step metabolic pathway engineering. |
Practical CRISPR/Cas9 Workflows for Metabolic Pathway Manipulation
Design Principles for sgRNAs Targeting Metabolic Genes and Enzymes
Within the broader thesis of applying CRISPR/Cas9 for metabolic engineering, the design of single guide RNAs (sgRNAs) is the most critical determinant of success. Targeting metabolic genes and enzymes presents unique challenges, including the need for precise allelic modulation, avoidance of compensatory pathway activation, and management of cellular fitness effects. This document outlines updated design principles, application notes, and protocols for creating high-efficiency, specific sgRNAs for metabolic engineering research and therapeutic target validation.
Modern sgRNA design integrates multiple predictive parameters. The following table synthesizes key metrics and their optimal ranges for targeting metabolic genes, based on current literature and algorithm outputs.
Table 1: Quantitative Parameters for Metabolic Gene sgRNA Design
| Parameter | Optimal Range/Target | Functional Rationale for Metabolic Targets |
|---|---|---|
| On-Target Efficiency Score | >70 (CHOPCHOP, Doench ‘16) | Ensures high probability of cutting, critical for polyploid genomes or high-copy number enzyme genes. |
| GC Content | 40-60% | Balances stability and unwinding efficiency; crucial for targeting GC-rich regulatory regions. |
| Specificity (Off-Target Score) | <50 potential off-targets (≤3 mismatches) | Vital to avoid unintended metabolic network perturbations and false phenotypes. |
| Seed Region Tm | High (>55°C) | Enhances on-target binding specificity, especially important for gene families with high homology (e.g., kinases, dehydrogenases). |
| 5' Terminal Nucleotide | G (for U6 promoter) | Maximizes transcription initiation; essential for consistent sgRNA expression in screening libraries. |
| Genomic Context | Exonic, early coding sequence | Promotes frameshift mutations and loss-of-function; avoid targeting functional domains if partial function is undesirable. |
| SNP Awareness | Check for variants in PAM/protospacer | Prevents failure in genetically diverse populations or specific cell lines. |
Protocol: Design and Validation of sgRNAs for a Metabolic Enzyme Gene
Objective: To design, clone, and validate sgRNAs targeting HMGCR (3-hydroxy-3-methylglutaryl-CoA reductase), a key enzyme in the cholesterol biosynthesis pathway.
Workflow Diagram Title: sgRNA Design & Validation Workflow
Materials & Reagents:
- Target Cells: HepG2 (human hepatoma) or relevant primary cells.
- Cloning Vector: lentiCRISPRv2 (Addgene #52961) or similar.
- Oligonucleotides: Designed sgRNA sequences synthesized as oligo duplexes.
- Enzymes: BsmBI-v2, T4 DNA Ligase, HiFi DNA Assembly Master Mix.
- Cell Culture Reagents: Polybrene (8 µg/mL), Puromycin (selection antibiotic).
- Validation Kits: HMGCR Activity Assay Kit (colorimetric), T7 Endonuclease I.
- Sequencing: Primers for genomic amplification of target locus.
Detailed Protocol:
Part A: sgRNA Design & Cloning
- Input the genomic sequence of human HMGCR (NCBI Reference Sequence) into the CHOPCHOP web tool.
- Filter outputs using the criteria in Table 1. Prioritize sgRNAs targeting the 5' region of the coding sequence within exon 4.
- Synthesize top 3-4 sgRNA oligos (forward: 5'-CACCG[N20]-3', reverse: 5'-AAAC[N20^C]-3').
- Digest the lentiCRISPRv2 vector with BsmBI for 1 hour at 55°C. Gel purify the linearized backbone.
- Anneal and phosphorylate oligo duplexes. Ligate into the digested vector using T4 DNA Ligase.
- Transform into Stbl3 competent cells. Sequence confirm with U6 forward primer.
Part B: Delivery & Selection
- Package lentivirus by co-transfecting each lentiCRISPRv2 sgRNA plasmid with psPAX2 and pMD2.G into Lenti-X 293T cells using PEI transfection reagent.
- Harvest virus supernatant at 48 and 72 hours post-transfection.
- Transduce HepG2 cells at ~30% confluence with virus plus 8 µg/mL Polybrene.
- Select transduced cells with 2-5 µg/mL puromycin beginning 48 hours post-transduction for 5-7 days.
Part C: Validation (Multi-Modal)
- Genotypic Validation (3 days post-selection):
- Extract genomic DNA from a portion of cells.
- PCR amplify the ~500bp region flanking the sgRNA target site.
- Hybridize and digest PCR products with T7EI. Analyze fragment cleavage on a 2% agarose gel to estimate indel efficiency. For precise quantification, submit PCR products for next-generation amplicon sequencing.
- Phenotypic & Metabolic Validation (7-10 days post-selection):
- Enzymatic Activity: Lyse cells and perform HMGCR activity assay per kit instructions. Normalize to total protein.
- Metabolomic Analysis: Extract lipids and analyze via LC-MS. Quantify downstream sterol intermediates (e.g., lanosterol, cholesterol) to confirm pathway disruption.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Metabolic Gene sgRNA Experiments
| Reagent/Material | Supplier Examples | Function in Protocol |
|---|---|---|
| lentiCRISPRv2 Vector | Addgene | All-in-one plasmid for constitutive expression of Cas9, sgRNA, and puromycin resistance. |
| BsmBI-v2 Restriction Enzyme | NEB | High-fidelity enzyme for precise digestion of the sgRNA scaffold cloning site. |
| Lenti-X 293T Cells | Takara Bio | High-titer lentiviral packaging cell line. |
| Transfection-Grade PEI | Polysciences | Cost-effective polymer for high-efficiency plasmid transfection into packaging cells. |
| Polybrene | Sigma-Aldrich | Cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. |
| T7 Endonuclease I | NEB | Detects mismatches in heteroduplex DNA, enabling rapid estimation of indel efficiency. |
| HMGCR Activity Assay Kit | Abcam / Sigma | Specific colorimetric assay to quantify the functional knockout of the target enzyme. |
| LC-MS Grade Solvents | Fisher Chemical | Essential for reproducible and high-sensitivity untargeted or targeted metabolomics. |
Pathway & Decision Logic Visualization
Metabolic Network Feedback Consideration: Knockout of a key enzyme (e.g., HMGCR) can activate feedback loops or alternative pathways. This logic must inform sgRNA selection and validation strategy.
Diagram Title: Metabolic Feedback & sgRNA Validation Logic
Conclusion: Effective metabolic gene targeting requires sgRNAs optimized not only for cutting efficiency but also for specificity within complex, interconnected genomes. A multi-modal validation protocol—combining genotypic, enzymatic, and metabolomic analyses—is essential to confirm knockout and understand resultant network adaptations. These principles and protocols provide a robust framework for advancing metabolic engineering and therapeutic discovery.
Application Notes
Within metabolic engineering research, the precise modification of industrial microbial and mammalian hosts using CRISPR/Cas9 necessitates efficient, scalable, and host-appropriate delivery systems. The choice between chemical transfection, viral vectors, and ribonucleoprotein (RNP) delivery critically impacts editing efficiency, cellular toxicity, laboratorial timelines, and regulatory compliance for therapeutic or bioproduction applications.
1. Chemical & Physical Transfection This method involves complexing nucleic acids (plasmid DNA or in vitro transcribed RNA) with cationic lipids or polymers, or using physical methods like electroporation to facilitate membrane passage. It is versatile and avoids viral safety concerns, making it suitable for early-stage research in various hosts. However, it often suffers from lower efficiency in hard-to-transfect industrial cell lines (e.g., CHO, primary cells), significant cytotoxicity, and the potential for genomic integration of plasmid DNA, which is undesirable for therapeutic cell line development.
2. Viral Vectors (Lentivirus & AAV) Viral vectors, particularly lentivirus (LV) and adeno-associated virus (AAV), offer high transduction efficiency across diverse cell types, including non-dividing cells. LVs enable stable genomic integration for persistent expression, useful for creating engineered cell pools. AAVs provide high-titer, transient expression with a favorable safety profile. In metabolic engineering, they are powerful for delivering large DNA donor templates for homology-directed repair (HDR). Drawbacks include limited cargo capacity, complex and costly GMP production, and immunogenicity concerns in clinical applications.
3. Ribonucleoprotein (RNP) Delivery Direct delivery of pre-assembled Cas9 protein and guide RNA as a complex represents the most rapid and precise method. RNPs act immediately upon delivery, minimizing off-target effects due to short intracellular persistence. This method is ideal for generating clonal cell lines with precise edits (knock-outs, small insertions) and is highly effective in hosts where nucleic acid delivery is inefficient. It avoids the need for host transcription/translation, reducing cell-type dependency. The primary challenge is delivery efficiency, often requiring specialized electroporation or microfluidics devices.
Quantitative Comparison of Delivery Systems
Table 1: Key Performance Metrics for CRISPR/Cas9 Delivery Systems in Industrial Hosts
| Parameter | Chemical Transfection (plasmid) | Viral Vector (Lentivirus) | RNP Delivery (Electroporation) |
|---|---|---|---|
| Typical Editing Efficiency (%) | 10-40% (highly variable) | 60-90% | 70-95% |
| Time to Genomic Edit (hrs) | 48-72 (requires transcription) | 48-72 (requires transduction) | 2-24 (immediate activity) |
| Cargo Capacity | High (>10 kb) | Limited (LV: ~8 kb, AAV: ~4.7 kb) | Very Limited (Cas9 protein + ~100 nt gRNA) |
| Integration Risk | Moderate (random integration) | High (LV) / Low (AAV) | None |
| Cytotoxicity | Moderate to High | Low to Moderate (immunogenicity) | Low |
| Protocol Complexity | Low | High (production & titration) | Medium (protein complexation) |
| Ideal Primary Use Case | Early-stage screening, easy-to-transfect lines | Stable cell line generation, hard-to-transfect cells | High-fidelity knock-outs, clinical applications |
Experimental Protocols
Protocol 1: Lipofection of CRISPR Plasmid DNA into CHO-K1 Cells for Metabolic Gene Knock-out Objective: To disrupt a gene in the cholesterol biosynthesis pathway in CHO cells using a plasmid expressing Cas9 and gRNA.
- Day 1: Seed CHO-K1 cells in a 24-well plate at 2.5 x 10^5 cells/well in CD CHO medium. Incubate at 37°C, 8% CO2.
- Day 2: Prepare transfection complexes. For each well, dilute 1 µg of px459 plasmid (encoding Cas9, gRNA, and Puromycin resistance) in 50 µL Opti-MEM. Dilute 2 µL of Lipofectamine 3000 reagent in 50 µL Opti-MEM. Incubate 5 min. Combine diluted DNA and reagent, mix, incubate 15 min at RT.
- Add complexes dropwise to cells. Rock plate gently.
- Day 3: (24h post-transfection) Begin selection with 5 µg/mL Puromycin. Maintain selection for 48-72 hours.
- Day 6: Split cells and allow recovery in standard medium. After 7 days, harvest genomic DNA and assess editing efficiency via T7 Endonuclease I assay or next-generation sequencing.
Protocol 2: Lentiviral Transduction for Stable gRNA Expression in HEK293T Cells Objective: To create a polyclonal cell population with stable integration of a gRNA targeting a glycolytic enzyme.
- Day 1: Seed HEK293T producer cells in a 6-well plate.
- Day 2: Co-transfect using PEI Max: 1 µg psPAX2 (packaging), 0.5 µg pMD2.G (VSV-G envelope), and 1.5 µg of lentiCRISPRv2 transfer plasmid (containing your gRNA). Change medium 6h post-transfection.
- Day 3 & 4: Harvest viral supernatant at 48h and 72h, filter through 0.45 µm PVDF filter, and store at 4°C or -80°C.
- Day 5: Transduce target HEK293 cells. Plate cells, then add viral supernatant supplemented with 8 µg/mL Polybrene. Spinfect at 800 x g for 30 min at 32°C. Return to incubator.
- Day 6: Replace with fresh medium.
- Day 8: Begin selection with 2 µg/mL Puromycin for 5-7 days to obtain a stable polyclonal pool for metabolic flux analysis.
Protocol 3: RNP Delivery via Neon Electroporation for Precise Editing in Primary T-Cells Objective: To knock-in a therapeutic transgene at a specific locus in human primary T-cells for immunotherapy research.
- Day 1: Isolate and activate primary human T-cells using CD3/CD28 Dynabeads and IL-2 for 48-72 hours.
- Day 3: Assemble RNP complex. Mix 60 pmol of purified Alt-R S.p. Cas9 V3 protein with 60 pmol of synthetic crRNA:tracrRNA duplex (Alt-R CRISPR-Cas9 system, IDT). Incubate at room temperature for 20 min.
- Prepare 10 µg of single-stranded DNA oligo donor (ssODN) for HDR.
- Wash 1 x 10^6 activated T-cells twice in PBS. Resuspend in "R" Buffer (Neon System) with RNP complex and ssODN.
- Electroporate using the Neon Transfection System (1400V, 10ms, 3 pulses).
- Immediately transfer cells to pre-warmed complete medium (IL-2 supplemented). Allow recovery for 48-72 hours before assessing editing efficiency via flow cytometry or sequencing.
Visualizations
Title: Decision Workflow for CRISPR Delivery Method Selection
Title: Intracellular Pathways: Viral Vector vs. RNP Delivery
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for CRISPR Delivery Experiments
| Reagent / Material | Supplier Examples | Function in Delivery Experiments |
|---|---|---|
| Lipofectamine 3000 | Thermo Fisher | Cationic lipid reagent for efficient plasmid or RNA transfection into mammalian cells. |
| Polyethylenimine (PEI Max) | Polysciences | High-efficiency, low-cost polymer for transfection of plasmid DNA, commonly used for viral vector production. |
| Lentiviral Packaging Mix | Takara, OriGene | Pre-mixed plasmids (psPAX2, pMD2.G) for simplified production of lentiviral particles. |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Tech. | High-purity, high-activity Cas9 protein optimized for RNP formation and delivery. |
| Neon Transfection System | Thermo Fisher | Electroporation device optimized for high-efficiency, low-toxicity delivery (esp. RNPs) into sensitive cells like primaries. |
| Polybrene | Sigma-Aldrich | Cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. |
| Opti-MEM Reduced Serum Media | Thermo Fisher | Low-serum medium used for diluting lipids/DNA during transfection complex formation, minimizing interference. |
| Puromycin Dihydrochloride | Thermo Fisher | Selection antibiotic for cells successfully transduced/transfected with constructs containing a puromycin resistance gene. |
Thesis Context: This application note details core CRISPR/Cas9 methodologies for achieving precise genomic alterations—knockouts, knock-ins, and multiplexed edits—within the framework of metabolic engineering research. These techniques enable the rational redesign of cellular metabolism for enhanced production of pharmaceuticals, biofuels, and fine chemicals.
Application Notes
Gene Knockouts for Eliminating Competing Pathways
In metabolic engineering, gene knockouts are essential to inactivate genes encoding enzymes in competing or regulatory pathways, thereby diverting metabolic flux toward a desired product. The double-strand break (DSB) generated by Cas9 is predominantly repaired by error-prone Non-Homologous End Joining (NHEJ), leading to small insertions or deletions (indels) that disrupt the coding sequence.
Key Quantitative Data: Table 1: Representative Knockout Efficiency Across Common Model Systems
| Organism/Cell Type | Target Gene | Delivery Method | Average Indel Efficiency (%) | Primary Readout |
|---|---|---|---|---|
| S. cerevisiae (Yeast) | PDC1 (Pyruvate Decarboxylase) | Plasmid (HR-based) | >95 | HPLC (Ethanol reduction) |
| HEK293T (Human) | HPRT1 (Housekeeping) | RNP (Electroporation) | 85-90 | Sanger Sequencing / SURVEYOR |
| CHO-K1 (Mammalian) | FUT8 (Fucosyltransferase) | Lentivirus | 70-80 | LC-MS (Glycan analysis) |
| E. coli | galK (Galactokinase) | Plasmid (λ-Red) | >99 | Phenotypic screening |
Precise Knock-ins for Pathway Insertion
Knock-ins facilitate the targeted insertion of heterologous metabolic pathway genes or regulatory elements via Homology-Directed Repair (HDR). This requires a donor DNA template with homology arms flanking the insert. In non-dividing cells or organisms with low HDR activity, strategies like using single-stranded donor oligonucleotides (ssODNs) or inhibiting NHEJ are employed.
Key Quantitative Data: Table 2: Knock-in Efficiency Variables
| Parameter | Typical Range/Choice | Impact on HDR Efficiency |
|---|---|---|
| Donor Template Form | dsDNA (plasmid), ssODN, AAV | ssODN: 0.1-5%; dsDNA: 1-20%; AAV: Can be higher |
| Homology Arm Length (each side) | 30-50 bp (ssODN), 500-1000 bp (dsDNA) | Longer arms generally increase efficiency |
| Cell Cycle Stage | S/G2 phase | Essential for HDR; synchronization can boost rates 2-4x |
| NHEJ Inhibition (e.g., Scr7) | 0-10 μM | Can increase HDR efficiency 1.5-3 fold |
| Cas9 Nickase (D10A) Use | Paired sgRNAs | Reduces indels at target, can improve precise integration |
Multiplexed Pathway Engineering
Multiplexed editing allows simultaneous knockout of several native genes and knock-in of multiple pathway components, enabling comprehensive pathway overhaul. This is achieved by co-expressing multiple single guide RNAs (sgRNAs) with Cas9 and appropriate donors.
Key Quantitative Data: Table 3: Outcomes of a Model Triplex Editing Experiment in Yeast for Terpenoid Production
| Target Locus | Edit Type | Donor | Editing Efficiency | Metabolic Outcome |
|---|---|---|---|---|
| ERG9 Promoter | Knock-down (Promoter Swap) | Weak promoter donor | 88% | Reduced ergosterol flux |
| ROX1 | Knockout | NHEJ-mediated | 92% | Derepression of aerobic genes |
| tHMGR | Knock-in (Genomic Integration) | Plasmid with pathway gene | 76% | Enhanced precursor supply |
| Combined | Multiplex | All components | Triple-positive: 41% | >50-fold product titer increase |
Detailed Protocols
Protocol 1: High-Efficiency Knockout via RNP Electroporation in Mammalian Cells
Aim: To disrupt the FUT8 gene in CHO-K1 cells to produce afucosylated antibodies with enhanced effector function.
Materials: See "The Scientist's Toolkit" below.
Method:
- sgRNA Design & Synthesis: Design a 20-nt guide sequence targeting an early exon of FUT8. Synthesize crRNA and tracrRNA separately or as a single guide RNA (sgRNA).
- RNP Complex Formation: Combine 10 μg of purified S. pyogenes Cas9 protein with 5 μg of sgRNA (molar ratio ~1:3). Incubate at 25°C for 10 minutes.
- Cell Preparation: Harvest 1x10^6 log-phase CHO-K1 cells, wash with PBS, and resuspend in 100 μL of electroporation buffer.
- Electroporation: Mix cell suspension with RNP complex. Transfer to a 2-mm cuvette and electroporate (e.g., 1350V, 30ms pulse length, 1 pulse). Immediately add pre-warmed medium.
- Recovery & Analysis: Plate cells and recover for 48-72 hours. Analyze editing efficiency via T7 Endonuclease I assay or ICE Analysis (Synthego) from genomic DNA. Clone cells by limiting dilution to isolate knockouts, confirmed by Sanger sequencing and LC-MS of antibody glycans.
Aim: To introduce a single amino acid change (R132H) in the IDH1 gene in HEK293T cells, a common mutation found in cancer metabolism studies.
Method:
- Design of Donor Template: Synthesize a 100-nt ssODN (ultramer) with the desired mutation centered, flanked by ~50-nt homology arms complementary to the target sequence. Phosphorothioate modifications on ends are recommended to stabilize the oligonucleotide.
- Co-delivery with CRISPR Components: Co-transfect 500 ng of Cas9 expression plasmid (or 10 μg of RNP), 250 ng of sgRNA expression plasmid (or 5 μg of in vitro transcribed sgRNA), and 1 μM of ssODN donor into HEK293T cells using a high-efficiency transfection reagent.
- HDR Enhancement: Treat cells with 5 μM Scr7, a DNA-PKcs inhibitor, or 2 mM Nocodazole for cell cycle synchronization at S/G2 phase, starting 6 hours post-transfection for 24-48 hours.
- Screening and Validation: After 72 hours, extract genomic DNA. Perform PCR amplification of the target locus and analyze via Sanger sequencing or next-generation sequencing (NGS) to quantify precise HDR efficiency. Isolate clones for downstream metabolic profiling (e.g., 2-HG production).
Protocol 3: Multiplexed Engineering Using a tRNA-sgRNA Array in Yeast
Aim: To simultaneously disrupt three genes (PDC1, ADH1, ALD6) and integrate a heterologous isobutanol production pathway in S. cerevisiae.
Method:
- Multiplex sgRNA Cassette Construction: Design four sgRNA sequences targeting the three knockout loci and one "safe harbor" integration site. Assemble these sequences into a single transcription unit on a plasmid, separating each sgRNA with a tRNA (e.g., tRNA^Gly) processing element using Golden Gate assembly. This creates a tRNA-sgRNA array.
- Donor DNA Construction: Assemble a single donor plasmid containing: a) Three repair templates for NHEJ-mediated knockout (short homology for promoter deletion), and b) A pathway expression cassette (e.g., kivD, ADH2) flanked by long homology arms (500 bp) for the "safe harbor" locus.
- Yeast Transformation: Co-transform the sgRNA array plasmid (expressing Cas9), the donor plasmid, and a marker selection plasmid into competent yeast cells using the lithium acetate method.
- Selection and Screening: Select on appropriate media. Screen colonies via multiplex colony PCR across all four target loci. Confirm pathway integration and gene disruptions by sequencing. Quantify isobutanol titer via GC-MS after fermentation in defined medium.
Visualizations
Diagram 1: Gene knockout via CRISPR-Cas9 and NHEJ
Diagram 2: Precise knock-in via HDR with donor template
Diagram 3: Multiplexed editing workflow for pathway engineering
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for CRISPR-based Metabolic Engineering
| Reagent/Material | Supplier Examples | Primary Function in Experiments |
|---|---|---|
| S. pyogenes Cas9 Nuclease (WT & D10A Nickase) | Integrated DNA Technologies (IDT), Thermo Fisher, Synthego | Creates targeted DSBs or nicks for genome editing. |
| Synthetic sgRNA (crRNA & tracrRNA) | IDT, Dharmacon, Synthego | Guides Cas9 to specific genomic DNA sequences. |
| Electroporation System (e.g., Neon, Nucleofector) | Thermo Fisher, Lonza | Enables high-efficiency RNP delivery into difficult cell types. |
| HDR Enhancers (SCR7, RS-1) | Sigma-Aldrich, Tocris | Small molecules that inhibit NHEJ or promote HDR, increasing knock-in efficiency. |
| Homology Donor Templates (ssODNs, gBlocks, AAV) | IDT, Genewiz, VectorBuilder | Provides repair template for precise HDR-mediated edits. |
| tRNA-sgRNA Cloning Kit | Addgene (Plasmid kits), NEB | Facilitates assembly of multiplex sgRNA expression arrays. |
| T7 Endonuclease I / Surveyor Nuclease | NEB, IDT | Detects indel mutations from NHEJ repair by cleaving mismatched heteroduplex DNA. |
| Next-Generation Sequencing (NGS) Kit for CRISPR | Illumina, Paragon Genomics | Provides deep, quantitative analysis of editing outcomes and off-target effects. |
Within the broader thesis on CRISPR/Cas9 for metabolic engineering, this document details the application of catalytically dead Cas9 (dCas9) as a programmable transcriptional regulator. By fusing dCas9 to effector domains, researchers can precisely activate (CRISPRa) or repress (CRISPRi) target genes without altering the DNA sequence. This approach is pivotal for dynamically rerouting metabolic fluxes in microbial and mammalian cell factories, enabling the optimized production of biofuels, pharmaceuticals, and commodity chemicals.
Key Principles & Quantitative Data
CRISPRa/i systems modulate transcription by targeting promoter or enhancer regions. The efficiency is influenced by guide RNA (sgRNA) design, effector domain strength, and genomic context.
Table 1: Common Effector Domains for dCas9-based Transcriptional Regulation
| Effector System | Core Domain(s) | Origin | Typical Target | Effect Size (Fold-Change)* | Key Applications in Metabolism |
|---|---|---|---|---|---|
| CRISPRi (Repression) | KRAB (Krüppel-associated box) | Mammalian | Promoter / TSS | 5-100x (repression) | Downregulation of competing pathways (e.g., byproduct formation) |
| CRISPRa (Activation) | VP64-p65-Rta (VPR) | Viral (HSV, etc.) | Promoter / Enhancer | 10-1000x (activation) | Upregulation of rate-limiting enzymes (e.g., in terpenoid pathways) |
| CRISPRa | SunTag + scFv-VP64 | Synthetic / Yeast | Promoter | 50-500x (activation) | Multigene activation for biosynthetic clusters |
| Synergistic Activation Mediator (SAM) | MS2-P65-HSF1 + dCas9-VP64 | Synthetic | Promoter / Gene Body | 100-1000x (activation) | High-level production of antibiotics in Streptomyces |
*Fold-change ranges are approximate and highly dependent on the specific gene and host organism. TSS: Transcription Start Site.
Table 2: Comparative Performance of CRISPRa/i in Model Organisms for Metabolic Engineering
| Host Organism | System | Target Pathway/ Gene | Metric | Result (vs. Wild-Type/Control) | Key Insight |
|---|---|---|---|---|---|
| E. coli | dCas9-VP64 (a) | gltA (TCA cycle) | Succinate Titer | 3.5-fold increase | Precise activation boosted flux through engineered branch. |
| S. cerevisiae | dCas9-KRAB (i) | FPS1 (farnesyl pyrophosphate shunt) | Amorphadiene Yield | 2.8-fold increase | Repression of competing pathway funneled flux toward desired product. |
| CHO Cells | SAM System (a) | Multiple genes in apoptosis pathway | Recombinant Protein Yield | 60% increase | Simultaneous activation of anti-apoptotic genes extended production phase. |
| B. subtilis | dCas9-VPR (a) | Acetoin biosynthetic genes | Acetoin Productivity | 4.1-fold increase | Activation of operon genes synergistically enhanced flux. |
Experimental Protocols
Protocol 1: Design and Cloning of dCas9-Effector and sgRNA Expression Constructs
Objective: To assemble plasmids expressing a dCas9-effector fusion and target-specific sgRNAs for metabolic pathway regulation.
Materials:
- Research Reagent Solutions: See "The Scientist's Toolkit" below.
- dCas9-VPR or dCas9-KRAB backbone plasmid (Addgene #xxx, #xxx).
- sgRNA scaffold plasmid (Addgene #xxx).
- Oligonucleotides for target-specific sgRNA (20nt guide sequence).
- Restriction enzymes (e.g., BsaI) and T4 DNA ligase.
- High-efficiency competent cells (e.g., NEB 5-alpha).
Methodology:
- sgRNA Design & Cloning: a. Design a 20-nucleotide guide sequence targeting the non-template strand of the promoter region (-50 to +100 relative to TSS) for activation or the promoter/TSS for repression. b. Synthesize complementary oligonucleotides with 5' overhangs compatible with your chosen sgRNA expression vector (e.g., for BsaI sites: Forward: 5'-CACCG[20nt guide]-3', Reverse: 5'-AAAC[20nt reverse complement]C-3'). c. Anneal oligos, phosphorylate, and ligate into the BsaI-digested sgRNA plasmid. d. Transform into competent E. coli, select on appropriate antibiotic, and sequence-verify clones.
- Delivery System Preparation: a. For simultaneous delivery, the dCas9-effector and sgRNA expression cassettes can be assembled into a single plasmid or kept separate. b. For multiplexing, clone multiple sgRNA arrays using Golden Gate assembly.
Protocol 2: Transfection and Screening in Mammalian Cell Culture (e.g., CHO cells)
Objective: To introduce CRISPRa/i components into mammalian production cell lines and screen for transcriptional changes and metabolic phenotypes.
Materials:
- CHO-S cells.
- Optimized growth medium.
- Lipofectamine 3000 or electroporation system.
- Puromycin or other appropriate selection antibiotic.
- qPCR reagents, RNA extraction kit.
Methodology:
- Transfection: a. Seed CHO-S cells in a 24-well plate 24h prior to reach 70-90% confluency. b. Co-transfect 500ng of dCas9-effector plasmid and 500ng of sgRNA plasmid(s) using Lipofectamine 3000 per manufacturer's protocol. c. Include controls: non-targeting sgRNA and untransfected cells.
- Selection & Screening: a. 48h post-transfection, begin selection with puromycin (e.g., 2 µg/mL) for 5-7 days to establish a polyclonal population. b. Harvest cells for analysis: - Transcriptional Analysis: Extract RNA, synthesize cDNA, perform qPCR for target metabolic genes and housekeeping controls. - Metabolite Analysis: Use HPLC or LC-MS to quantify extracellular/intracellular metabolites (e.g., product titers, pathway intermediates). - Growth Phenotype: Monitor cell density and viability to assess burden or beneficial effects.
Protocol 3: Metabolic Flux Analysis in Yeast via CRISPRi
Objective: To quantify the redistribution of central carbon flux upon targeted gene repression in S. cerevisiae.
Materials:
- Yeast strain with integrated dCas9-KRAB.
- sgRNA expression plasmid (yeast 2µ origin, URA3 marker).
- ( ^{13}\text{C} )-labeled glucose (e.g., [1-( ^{13}\text{C} )]glucose).
- GC-MS system.
- YPD and synthetic dropout media.
Methodology:
- Strain Preparation: Transform the sgRNA plasmid into the dCas9-KRAB yeast strain and select on -Ura plates.
- ( ^{13}\text{C} ) Tracer Experiment: a. Inoculate single colonies into -Ura medium with unlabeled glucose and grow overnight. b. Subculture into fresh medium containing the ( ^{13}\text{C} )-labeled glucose at mid-exponential phase. c. Harvest cells during exponential growth phase (OD~5) by rapid filtration.
- Metabolite Extraction & Analysis: a. Quench metabolism with cold methanol, perform intracellular metabolite extraction. b. Derivatize metabolites (e.g., amino acids, organic acids) for GC-MS analysis. c. Measure mass isotopomer distributions (MIDs) of key metabolites from the target pathway (e.g., TCA cycle, glycolysis).
- Flux Calculation: Use computational software (e.g., INCA, ( ^{13}\text{C} )FLUX2) to fit the MIDs to a metabolic network model and estimate in vivo reaction fluxes. Compare fluxes between the CRISPRi strain and a non-targeting sgRNA control.
Visualizations
Title: Mechanism of CRISPRa and CRISPRi Transcriptional Control
Title: CRISPRa/i Workflow for Metabolic Flux Engineering
Title: Example: Rewiring Flux in a Polyketide Pathway
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application | Example/Supplier |
|---|---|---|
| dCas9-Effector Plasmids | Backbone vectors expressing dCas9 fused to activator (VPR, p65-HSF1) or repressor (KRAB) domains. Essential for system function. | Addgene: pAC1542 (dCas9-VPR), pAN1511 (dCas9-KRAB). |
| sgRNA Cloning Vectors | Plasmids containing the sgRNA scaffold for easy insertion of target-specific 20nt guide sequences via Golden Gate or BbsI/BsaI cloning. | Addgene: pAN1582 (for mammalian), pMLS261 (for yeast). |
| CRISPRa/i Library | Pooled collections of sgRNAs targeting entire gene families (e.g., all kinases, metabolic enzymes). For high-throughput screening of flux determinants. | Custom libraries from Twist Bioscience; pre-made from suppliers like Dharmacon. |
| Lipofectamine 3000 | A cationic lipid-based transfection reagent for high-efficiency delivery of plasmid DNA into a wide range of mammalian cell lines, including CHO and HEK293. | Thermo Fisher Scientific, Cat. No. L3000015. |
| Puromycin Dihydrochloride | A selective antibiotic for mammalian cells. Used to select for cells that have stably integrated or maintain plasmids carrying the puromycin resistance gene. | Thermo Fisher Scientific, Cat. No. A1113803. |
| [1-13C]Glucose | Isotopically labeled carbon source for 13C Metabolic Flux Analysis (13C-MFA). Enables precise quantification of intracellular metabolic reaction rates. | Cambridge Isotope Laboratories, CLM-1396. |
| RNeasy Kit | For rapid, high-quality total RNA purification from bacterial, yeast, or animal cells. Critical for downstream qPCR validation of transcriptional changes. | Qiagen, Cat. No. 74104. |
| HPLC/MS-Grade Solvents | High-purity solvents (acetonitrile, methanol, water) for metabolite extraction and LC-MS analysis to quantify pathway intermediates and products. | Fisher Chemical, Optima LC/MS Grade. |
This application note details CRISPR/Cas9-mediated metabolic engineering in three chassis organisms. It supports the broader thesis that CRISPR/Cas9 is a transformative tool for redirecting cellular metabolism for industrial production. Protocols are optimized for researchers and industry scientists.
Case Study 1: EngineeringS. cerevisiaefor Bioisoprenoid Production
Application Note: This study aimed to increase farnesene yield, a precursor to biofuels and squalene (pharmaceutical adjuvant), by overexpressing the mevalonate (MVA) pathway and disrupting competing pathways.
Key Quantitative Data:
Table 1: Metabolic Engineering Impact on Farnesene Titers in S. cerevisiae
| Strain Description | Key Genetic Modification(s) | Cultivation Time (h) | Final Titer (g/L) | Yield (g/g glucose) | Reference/Year |
|---|---|---|---|---|---|
| Wild-type Control | None | 120 | 0.01 | 0.0002 | Baseline |
| MVA Overexpression | tHMG1 overexpression, ERG9 promoter down-tuning | 120 | 1.8 | 0.045 | (Dai et al., 2023) |
| CRISPR-Engineered | Cas9-mediated ERG9 repression, IDI1, ERG20 integration at ho locus | 96 | 2.5 | 0.062 | Current Best Practice |
Protocol: CRISPR/Cas9-Mediated Gene Integration and Repression in Yeast
- gRNA Design & Plasmid Construction: Design two 20-nt gRNAs targeting the ho locus ("safe harbor") and the promoter region of ERG9 (squalene synthase). Clone them into plasmid pCRCT (Addgene #126079) expressing Cas9 and a donor DNA template containing IDI1 and ERG20 under strong promoters.
- Yeast Transformation: Transform log-phase S. cerevisiae CEN.PK2 using the LiAc/SS Carrier DNA/PEG method. Plate on synthetic complete media lacking uracil for selection.
- Screening & Validation: Pick 10-15 colonies. Verify integration at the ho locus via colony PCR using flanking primers. Check ERG9 promoter editing by sequencing. Measure farnesene titers in 5 mL micro-cultures using GC-MS.
- Fermentation: Scale engineered strain in a 2 L bioreactor with defined mineral media, 2% glucose feed. Maintain pH 6.5, 30°C, 500 rpm agitation. Sample every 24h for product and substrate analysis.
Research Reagent Solutions for Yeast Engineering:
| Reagent/Material | Function | Example Product/Cat. No. |
|---|---|---|
| pCRCT Plasmid | All-in-one CRISPR/Cas9 expression and donor template cloning for yeast. | Addgene #126079 |
| Yeast Synthetic Drop-out Medium | Selective growth of transformants. | MilliporeSigma Y1501 |
| Zymolyase | Digests yeast cell wall for genomic DNA extraction. | Fujifilm 07665-55 |
| Farnesene Standard | Quantification standard for GC-MS calibration. | MilliporeSigma W374708 |
| Toolkit Table Reference: Key materials for replicating the yeast farnesene production protocol. |
Case Study 2: EngineeringE. colifor Polyketide Synthesis
Application Note: This protocol describes refactoring the E. coli genome for deoxyerythronolide B (DEB) production, a polyketide precursor to antibiotics like erythromycin.
Key Quantitative Data:
Table 2: DEB Production in Engineered E. coli Strains
| Strain/Intervention | Genetic Target(s) | Cultivation Vessel | Max Titer (mg/L) | Productivity (mg/L/h) |
|---|---|---|---|---|
| PLASMID-BASED (pSGP) | DEBS genes on plasmid | Shake Flask | 12 | 0.25 |
| CRISPR-CHROMOSOMAL | Cas9-assisted insertion of 30-kb DEBS cluster at attTn7 | Shake Flask | 78 | 1.63 |
| CRISPR-CHROMOSOMAL | As above + sfp (phosphopantetheinyl transferase) integration | Fed-Batch Bioreactor | 1100 | 15.3 |
Protocol: CRISPR/Cas9-Mediated Large Pathway Integration in E. coli
- Lambda Red & Cas9 Preparation: Transform the production strain with pKD46 (Lambda Red, temperature-sensitive) and pCas9cr4 (constitutively expresses Cas9). Grow at 30°C with antibiotics.
- Linear Donor & gRNA Construction: PCR-amplify the 30-kb DEBS polyketide synthase gene cluster with 500-bp homology arms for the attTn7 site. In vitro, transcribe a gRNA targeting attTn7.
- Electroporation: Induce Lambda Red with L-arabinose. Electroporate 100 µL competent cells with 500 ng linear donor and 200 ng gRNA. Recover in SOC at 30°C for 3h.
- Curing Plasmids: Heat-shock culture at 42°C to cure pKD46. Use plasmid incompatibility to cure pCas9cr4.
- Analytical: Confirm integration by long-read sequencing. Quantify DEB via LC-MS using extracted ion chromatogram for m/z 393.2.
Research Reagent Solutions for E. coli Engineering:
| Reagent/Material | Function | Example Product/Cat. No. |
|---|---|---|
| pCas9cr4 Plasmid | Constitutively expresses Cas9 for genome editing in E. coli. | Addgene #62655 |
| pKD46 Plasmid | Expresses Lambda Red recombinase under arabinose-inducible promoter. | Addgene #60609 |
| GeneArt Strings DNA Fragments | Custom, long linear donor DNA with homology arms. | Thermo Fisher Scientific |
| DEB (6-dEB) Standard | LC-MS standard for polyketide quantification. | Sigma-Aldrieb D5695 |
Case Study 3: Engineering CHO Cells for Monoclonal Antibody (mAb) Yield
Application Note: This protocol uses CRISPRi (CRISPR interference) to silence genes inhibiting apoptosis and increase cell longevity in fed-batch culture, thereby boosting mAb titers.
Key Quantitative Data:
Table 3: Impact of Anti-Apoptotic Engineering on CHO Cell Performance
| Cell Line / Intervention | Target Gene(s) (CRISPRi) | Viable Cell Density (10^6 cells/mL) | Viability >80% (Days) | Final mAb Titer (g/L) | Increase vs. Parent |
|---|---|---|---|---|---|
| CHO-S Parental | None | 8.2 | 7 | 2.1 | Baseline |
| Engineered Pool | BAX, CASP3 | 10.5 | 9 | 3.0 | +43% |
| Engineered Clone | BAX, CASP3, CASP7 | 12.1 | 12 | 3.8 | +81% |
Protocol: CRISPRi-Mediated Gene Repression in CHO Cells for Enhanced Production
- dCas9-KRAB & gRNA Lentivirus Production: Co-transfect HEK293T cells with packaging plasmids (psPAX2, pMD2.G) and either pLV-dCas9-KRAB (Addgene #99373) or pLV-U6-sgRNA-EF1a-Puro vectors containing gRNAs against BAX, CASP3, CASP7. Harvest virus at 48h and 72h.
- CHO Cell Transduction & Selection: Transduce CHO-S cells (in CD CHO medium + 8 µg/mL polybrene) with dCas9-KRAB virus first. Select with blasticidin (10 µg/mL) for 10 days. Subsequently, transduce with pooled sgRNA viruses and select with puromycin (5 µg/mL).
- Clone Isolation & Screening: Perform limiting dilution cloning. Screen ~100 clones via CellTiter-Glo for viable cell density at day 7 of batch culture. Top 10 clones are assessed for target gene knockdown via qRT-PCR.
- Fed-Batch Bioreactor Run: Cultivate top clone in 5 L bioreactor with CD CHO, 36°C, pH 7.0, 30% DO. Feed with EfficientFeed C+ starting day 3. Measure mAb titer daily via Protein A HPLC.
Research Reagent Solutions for Mammalian Cell Engineering:
| Reagent/Material | Function | Example Product/Cat. No. |
|---|---|---|
| pLV-dCas9-KRAB | Lentiviral vector for stable dCas9-KRAB (CRISPRi) expression. | Addgene #99373 |
| psPAX2 & pMD2.G | 2nd/3rd gen lentiviral packaging plasmids. | Addgene #12260 & #12259 |
| CD CHO Medium | Chemically defined, protein-free medium for CHO cell culture. | Gibco 10743029 |
| CellTiter-Glo 2.0 | Luminescent assay for quantifying viable cells. | Promega G9242 |
Solving CRISPR/Cas9 Challenges: Maximizing Efficiency and Specificity in Metabolic Hosts
Identifying and Mitigating Off-Target Effects in Complex Genomes
Within the thesis on CRISPR/Cas9 for metabolic engineering, the precision of genome editing is paramount. Off-target effects—unintended modifications at genomic loci with sequence similarity to the target site—pose a significant risk, potentially disrupting native metabolic pathways or causing cellular toxicity. This document provides application notes and detailed protocols for identifying and mitigating these effects in complex eukaryotic genomes, such as those of industrially relevant yeast, fungi, and mammalian cell lines used in metabolic engineering.
The following table summarizes the core characteristics of current methodologies.
Table 1: Comparison of Key Off-Target Identification and Validation Methods
| Method | Principle | Throughput | Detection Limit (Indel Frequency) | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| In Silico Prediction (e.g., CRISPOR, ChopChop) | Algorithmic search for genomic sites with homology to sgRNA. | Very High | N/A (Predictive) | Fast, cost-effective initial guide screening. | High false-negative rate; misses structurally accessible sites. |
| CIRCLE-Seq | In vitro cleavage of circularized genomic DNA followed by high-throughput sequencing. | High | ~0.01% | Highly sensitive; cell-free reduces bias. | Does not account for cellular chromatin context. |
| GUIDE-Seq | Integration of dsODN tags into double-strand breaks in vivo, followed by sequencing. | Medium-High | ~0.01% | Unbiased discovery in living cells. | Requires dsODN transfection, which can be cytotoxic. |
| SITE-Seq | In vitro cleavage of chromatin-associated DNA, capturing chromatin accessibility. | High | ~0.1% | Incorporates biochemical chromatin accessibility. | In vitro system; more complex protocol. |
| WGS (Whole Genome Sequencing) | Sequencing of entire edited genome to identify all variants. | Low | ~5% (practical) | Truly genome-wide, hypothesis-free. | Very costly; low sensitivity for rare indels; high data burden. |
| Targeted Amplicon Sequencing | Deep sequencing of PCR amplicons spanning predicted off-target loci. | Medium | ~0.1% | Cost-effective, highly sensitive validation. | Requires prior knowledge of loci to interrogate. |
Detailed Protocols
Protocol 3.1: Integrated Workflow for Off-Target Assessment in Metabolic Engineering Strains
Application: For comprehensive off-target profiling of a lead sgRNA designed to integrate a metabolic pathway gene into a fungal genome.
I. Materials & Reagents
- Fungal strain (e.g., S. cerevisiae, Y. lipolytica)
- CRISPR/Cas9 plasmid (e.g., with SpCas9 and sgRNA expression cassettes)
- HDR template DNA for metabolic gene integration
- CIRCLE-Seq Kit (e.g., ICE-seq)
- GUIDE-Seq dsODN (for mammalian cells; adapted protocols for fungi exist)
- NGS library preparation kits
- PCR reagents and primers for targeted amplicon sequencing
- Bioinformatics software: CRISPOR, Cas-OFFinder, BWA, CRISPResso2
II. Procedure
- Step 1: In Silico Screening. Input your 20-nt sgRNA sequence into CRISPOR. Compile a list of top 20-50 predicted off-target sites (allowing up to 4 mismatches, include bulges).
- Step 2: Primary Editing & Validation. Transfer CRISPR system and HDR template into your strain. Confirm on-target integration via diagnostic PCR and phenotypic assay (e.g., metabolite production).
- Step 3: Unbiased Discovery (CIRCLE-Seq).
- Isolate genomic DNA from a pool of edited clones.
- Shear and circularize DNA.
- In vitro digest with purified Cas9:sgRNA ribonucleoprotein (RNP).
- Linearize cleaved circles, add sequencing adapters, and perform NGS.
- Bioinformatics pipeline: Map reads to reference genome, identify breakpoint junctions.
- Step 4: Candidate Locus Validation.
- Merge predicted (Step 1) and discovered (Step 3) off-target loci.
- Design PCR primers to generate ~300 bp amplicons spanning each locus.
- Perform deep amplicon sequencing (20,000x coverage minimum) on genomic DNA from edited and wild-type control pools.
- Analyze with CRISPResso2 to quantify indel frequencies at each locus.
- Step 5: Mitigation & Re-design. If concerning off-target activity (>0.5% indel frequency in a coding or regulatory region) is detected, employ mitigation strategies (see Section 4) and repeat validation with a new sgRNA design.
Protocol 3.2: High-Fidelity Cas Variant Validation Protocol
Application: Comparing off-target profiles of WT SpCas9 to high-fidelity variant SpCas9-HF1 when using a sgRNA for activating a key enzyme promoter.
I. Materials
- HEK293T cells (or relevant mammalian cell line for metabolic engineering)
- Plasmids: pX330 (WT SpCas9), pX330-HF1 (SpCas9-HF1), identical sgRNA expression vector.
- GUIDE-Seq reagents.
- Flow cytometry sorter (if using FACS-based enrichment).
II. Procedure
- Co-transfection: Co-transfect HEK293T cells in triplicate with (a) WT Cas9 + sgRNA + GUIDE-Seq dsODN, or (b) HF1 Cas9 + sgRNA + GUIDE-Seq dsODN.
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract high-molecular-weight genomic DNA.
- GUIDE-Seq Library Preparation:
- Fragment DNA by sonication.
- Enrich for dsODN-tagged fragments using PCR with a biotinylated primer complementary to the dsODN.
- Prepare NGS libraries from enriched fragments.
- Sequencing & Analysis:
- Perform paired-end sequencing.
- Process data using the standard GUIDE-Seq computational pipeline.
- Compare the number and indel frequencies of off-target sites identified for WT Cas9 vs. HF1 Cas9.
- Phenotypic Correlation: Assess the on-target editing efficiency (via T7EI or sequencing) and the resulting transcriptional activation of the target promoter (via qRT-PCR) for both systems to confirm maintained on-target efficacy.
Mitigation Strategies: A Decision Guide
Table 2: Off-Target Mitigation Strategies and Their Applications
| Strategy | Mechanism | Best Use Case | Considerations for Metabolic Engineering |
|---|---|---|---|
| High-Fidelity Cas Variants (e.g., SpCas9-HF1, eSpCas9) | Engineered to reduce non-specific DNA contacts. | First choice for most new designs. | Verify on-target efficiency remains high for your specific genomic locus. |
| Truncated sgRNAs (tru-gRNAs) | Shorter guide (17-18 nt) reduces stability of off-target binding. | When high-fidelity variants show reduced on-target activity. | Can be combined with high-fidelity variants. Requires empirical testing. |
| RiboNucleoProtein (RNP) Delivery | Short-lived activity of pre-formed Cas9:sgRNA complex. | Transient editing; preferred for protoplast/fungal editing. | Reduces prolonged exposure and potential off-target cleavage. |
| Modified sgRNA Scaffolds (e.g., x- or e-sgRNA) | Alters scaffold structure to favor on-target conformation. | When standard guides show high off-target propensity. | Compatibility with chosen Cas variant must be confirmed. |
| Anti-CRISPR Proteins (AcrIIA4) | Inhibits Cas9 activity after a defined editing window. | For precise temporal control in inducible systems. | Emerging technology; requires careful dosing and delivery. |
| Computational sgRNA Design | Selects guides with unique sequences in the genome. | Foundational step for all experiments. | Use multiple prediction tools and prioritize guides with minimal high-similarity off-targets. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Analysis
| Item | Function & Application | Example Product/Provider |
|---|---|---|
| High-Fidelity Cas9 Expression Plasmid | Provides the nuclease with reduced off-target activity. | Addgene: pX330-HF1 (SpCas9-HF1). |
| CIRCLE-Seq Kit | All-in-one reagent kit for sensitive, cell-free off-target discovery. | ICE-seq Kit (ToolGen). |
| GUIDE-Seq dsODN | Double-stranded oligodeoxynucleotide tag for in vivo off-target capture. | Alt-R GUIDE-Seq Oligo (IDT). |
| Next-Generation Sequencing Service | For deep amplicon sequencing of validated off-target loci. | Illumina MiSeq, Amplicon-EZ (Genewiz). |
| CRISPResso2 Analysis Software | Quantifies indel frequencies from amplicon sequencing data. | Open-source web tool or standalone package. |
| Alt-R S.p. Cas9 Nuclease V3 | High-purity, recombinant Cas9 for RNP formation and in vitro assays. | Integrated DNA Technologies (IDT). |
| Genomic DNA Isolation Kit | For high-quality, high-molecular-weight DNA from edited cells. | DNeasy Blood & Tissue Kit (Qiagen). |
| Off-Target Prediction Web Tool | Free, comprehensive algorithm for initial sgRNA design and risk assessment. | CRISPOR (crispor.tefor.net). |
Visualizations
Diagram 1: Off Target Analysis Workflow
Diagram 2: Mitigation Strategy Decision Tree
Within the broader thesis on leveraging CRISPR/Cas9 for metabolic engineering, a central bottleneck is the reliable insertion of large DNA cargoes via Homology-Directed Repair (HDR). While effective in dividing mammalian cells or model microbes like S. cerevisiae, HDR is inherently inefficient in non-dividing cells (e.g., stationary-phase industrial bacteria, primary human T-cells for immunotherapy) and in many industrially relevant microbes (e.g., Corynebacterium, Streptomyces, non-model cyanobacteria) due to low native homologous recombination (HR) machinery activity. This application note details current strategies and protocols to enhance HDR efficiency in these challenging hosts, enabling precise metabolic pathway integration.
The following table summarizes key strategies, their mechanisms, and representative quantitative outcomes from recent literature (2023-2024).
Table 1: Comparative Strategies for Enhancing HDR Efficiency
| Strategy Category | Specific Approach | Target Organism | Reported HDR Efficiency Increase (vs. Baseline) | Key Mechanism |
|---|---|---|---|---|
| Host Machinery Modulation | Constitutive expression of phage-derived single-stranded annealing proteins (SSAPs) like RecT or GP35. | E. coli (stationary phase), Pseudomonas putida | 5 to 15-fold (up to ~40% absolute efficiency) | SSAPs catalyze recombination of single-stranded DNA, bypassing canonical RecA-dependent pathways. |
| Inducible expression of endogenous HR proteins (e.g., RecBCD, RecA, RecF). | Corynebacterium glutamicum, Synechococcus spp. | 3 to 8-fold | Boosts the cellular concentration of the native repair machinery. | |
| Repair Pathway Engineering | CRISPR/Cas9 nickase (nCas9) paired with SSAPs. | Primary human T-cells (non-dividing) | ~2 to 5-fold (reaching ~30% in some studies) | nCas9 creates single-strand breaks, reducing toxic indels and providing a preferred substrate for SSAPs. |
| Inhibition of Non-Homologous End Joining (NHEJ) via small molecules (e.g., Scr7) or genetic knockout of ligD or ku genes. | Aspergillus niger, Yarrowia lipolytica | 4 to 10-fold (NHEJ-deficient strains) | Shunts DNA repair away from error-prone NHEJ and toward HR/HDR pathways. | |
| Donor DNA Optimization | Use of single-stranded DNA (ssDNA) donors (100-200 nt) with 40-60 bp homologies. | E. coli, Bacillus subtilis | Up to 65% absolute efficiency in dividing cells; significant gains in non-dividers | SSDNA is a direct substrate for SSAPs and avoids transcription/replication conflicts. |
| Concatemerized double-stranded DNA (dsDNA) donors delivered on plasmids or as linear fragments. | Streptomyces coelicolor | ~50% efficiency for gene insertions | Increases local donor concentration and effective homology length. | |
| Cell Cycle & State Synchronization | Transient induction of competence programs (e.g., com genes in Bacillus). | B. subtilis (stationary phase) | ~1000-fold over non-competent cells | Artificially induces a state with active DNA uptake and recombination. |
| Chemical treatment (e.g., nocodazole) to arrest eukaryotic cells in S/G2 phase. | Primary fibroblasts | ~3-fold increase | Restricts HDR to cell cycle phases where homologous recombination is naturally active. |
Detailed Experimental Protocols
Protocol 1: HDR in Non-DividingE. coliusing ssDNA Donors and RecT Expression
Objective: Precise point mutation or small tag insertion in stationary-phase E. coli cells. Key Reagents: pCas9cr4 plasmid (or similar, expressing Cas9, sgRNA, and λ-Red/RecT), chemically synthesized ssDNA donor oligo.
- Strain Preparation: Transform your target E. coli strain with the pCas9cr4 plasmid. Select on appropriate antibiotics.
- Donor Design: Synthesize an ssDNA oligo (sense or antisense strand) containing the desired edit flanked by 50-60 bp homology arms. Phosphorothioate modifications at terminal 3-4 bases enhance stability.
- Induction of Repair Proteins: Inoculate a single colony into medium with antibiotic and 0.2% L-arabinose to induce λ-Red/RecT proteins. Grow to mid-log phase (OD600 ~0.5).
- Electrocompetent Cell Preparation: Chill culture on ice, wash 3x with ice-cold 10% glycerol. Concentrate cells 100x.
- Electroporation: Mix 50 µL cells with 100-200 ng of pSG (sgRNA plasmid targeting your locus) and 1 µM (final) of ssDNA donor oligo. Electroporate (1.8 kV, 200Ω, 25µF for 0.1 cm cuvette). Immediately recover in 1 mL SOC for 1-2 hours at 30°C.
- Counter-Selection & Screening: Plate on antibiotic selecting for the edit (if applicable) and incubate at 30°C. Screen colonies via colony PCR and Sanger sequencing.
Protocol 2: NHEJ Inhibition to Boost HDR in a Fungal Host (Aspergillus niger)
Objective: Gene knock-in via homologous recombination in a wild-type NHEJ-proficient strain. Key Reagents: CRISPR/Cas9 plasmid, dsDNA donor with >1 kb homologies, Scr7 (NHEJ inhibitor).
- Fungal Protoplast Preparation: Grow A. niger spores in liquid culture for 16-20 h. Harvest mycelia, digest with lysing enzymes (e.g., 10 mg/mL Lysing Enzymes from Trichoderma harzianum) in osmotic stabilizer (1.2 M MgSO4) for 3-4 h at 30°C. Filter, wash, and resuspend protoplasts in STC buffer.
- CRISPR/Cas9 & Donor Delivery: Co-transform 10^7 protoplasts with 5 µg of Cas9-sgRNA expressing plasmid and 1 µg of linear dsDNA donor fragment using PEG-mediated transformation.
- NHEJ Inhibition: Add Scr7 (final conc. 5-10 µM) directly to the transformation mixture and to the regeneration agar plates.
- Regeneration & Selection: Plate protoplasts on osmotically stable regeneration agar with Scr7. Incubate at 30°C for 3-5 days until transformant colonies appear.
- Genotyping: Pick colonies, isolate genomic DNA, and confirm correct integration by diagnostic PCR and Southern blot.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for HDR Enhancement Experiments
| Item | Function/Description | Example Vendor/Catalog |
|---|---|---|
| Phage-derived SSAP Expression Plasmids | Constitutively or inducibly express RecT, GP35, or other SSAPs to provide orthogonal recombination machinery. | Addgene (#62225, pSIM series; #166598, pCOEdT). |
| NHEJ Inhibitor (Scr7) | Small molecule inhibitor of DNA Ligase IV, temporarily suppresses the competing NHEJ pathway in eukaryotes and some fungi. | Sigma-Aldrich (SML1546). |
| Chemically Modified ssDNA Donors | Ultramer or PAGE-purified ssDNA oligos with phosphorothioate bonds for enhanced nuclease resistance. | Integrated DNA Technologies (IDT). |
| nCas9 (D10A) Nickase Plasmids | For generating single-strand breaks, reducing off-target effects and favoring HDR over NHEJ. | Addgene (#41816, pX335). |
| Cell Cycle Synchronization Agents | Nocodazole or Aphidicolin to arrest cultured eukaryotic cells in mitosis or S-phase, respectively, promoting HDR. | Cayman Chemical (13857, 14217). |
| Gibson or HiFi DNA Assembly Master Mix | For rapid and seamless assembly of concatemeric or complex dsDNA donor constructs. | New England Biolabs (E2611, E2621). |
| Protoplast Generation Kit (Fungal) | Standardized enzyme mixtures for reliable generation of fungal protoplasts for transformation. | Sigma-Aldrich (L1412). |
Visualization Diagrams
Title: Strategic Workflow for Enhancing HDR Efficiency
Title: Competing DNA Repair Pathways After CRISPR Cut
Overcoming Cellular Toxicity and Delivery Barriers in Refractory Organisms
Within a broader thesis on CRISPR/Cas9 for metabolic engineering, a critical roadblock is the application of these tools to non-model, industrially relevant refractory organisms. These organisms (e.g., extremophiles, non-conventional yeasts, certain anaerobic bacteria) often possess innate barriers such as robust cell walls, efficient efflux pumps, restriction-modification systems, and lack of established genetic tools. Furthermore, constitutive expression of CRISPR components, particularly Cas9, can induce cellular toxicity and DNA damage responses, stalling growth and killing cells before editing occurs. This Application Note details protocols and strategies to overcome these specific challenges.
Table 1: Common Barriers in Refractory Organisms and Their Impact
| Barrier Type | Example Organisms | Consequence for CRISPR Editing | Typical Success Rate (Untreated) |
|---|---|---|---|
| Physical Delivery | Mycobacteria, Microalgae, Filamentous Fungi | Impeded macromolecule entry; PEG-mediated transfection inefficient. | 1-5% transformation efficiency |
| Cellular Toxicity | Anaerobes (e.g., Clostridium), Primary T-cells | Constitutive Cas9 expression triggers SOS/p53 response; cell death. | <1% viable edited colonies |
| Restriction Systems | Wild-type Bacillus strains, Cyanobacteria | Foreign DNA (plasmids) degraded upon entry. | 0.01-0.1% transformation efficiency |
| Expression & Fidelity | Archaea, Acidophiles | Host polymerases/RNaases fail to process standard expression constructs. | Variable, often low editing fidelity |
Table 2: Strategies to Mitigate Toxicity and Improve Delivery
| Strategy | Mechanism | Resultant Improvement (Typical Range) |
|---|---|---|
| Ribonucleoprotein (RNP) Delivery | Direct delivery of pre-complexed Cas9 protein and sgRNA. | Reduces toxicity; increases speed. Efficiency gains: 5-50x over plasmid. |
| Inducible/Transient Expression | Use of tightly regulated promoters (e.g., anhydrotetracycline). | Limits Cas9 exposure. Viability increase: 10-100x. |
| Cell Wall Weakening | Pre-treatment with sub-inhibitory antibiotics (e.g., glycine, penicillin). | Increases permeability. Transformation boost: 3-20x. |
| Vector Modification | Use of host-derived replicons; methylation of plasmids in vitro. | Evades restriction systems. Efficiency increase: 10-1000x. |
Detailed Experimental Protocols
Protocol 3.1: RNP Delivery via Electroporation for Toxicity-Prone Bacteria
Objective: To achieve high-efficiency gene knockout in a refractory Gram-positive bacterium (Clostridium thermocellum) with minimal toxicity. Materials: Bacterial culture, custom sgRNA (chemically modified), purified S. pyogenes Cas9 protein, electroporation cuvettes (2 mm gap), Gene Pulser, recovery medium. Procedure:
- sgRNA Preparation: Resuspend synthetic sgRNA in nuclease-free buffer. Anneal tracerRNA and crRNA if using a dual-RNA system.
- RNP Complex Formation: Mix 100 pmol of Cas9 protein with 120 pmol of sgRNA in a total volume of 10 µL. Incubate at 25°C for 10 minutes.
- Cell Preparation: Grow culture to mid-log phase (OD600 ~0.5). Chill on ice for 15 min. Pellet cells (5,000 x g, 10 min, 4°C). Wash 3x with ice-cold 1 mM HEPES buffer (pH 7.0) containing 300 mM sucrose. Resuspend in wash buffer at 100x concentration.
- Electroporation: Combine 50 µL cell suspension with 10 µL RNP complex. Transfer to pre-chilled electroporation cuvette. Apply pulse (e.g., 1.8 kV, 600 Ω, 25 µF for C. thermocellum). Immediately add 1 mL pre-warmed recovery medium.
- Recovery and Plating: Transfer to anaerobic chamber. Recover for 4-6 hours at 37°C without selection. Plate on selective medium and incubate anaerobically for 48-72 hours. Screen colonies via PCR and sequencing.
Protocol 3.2: Plasmid Delivery Optimization Using Methylation and Cell Wall Weakening
Objective: To transform a wild-type Bacillus subtilis strain with a high-restriction activity. Materials: Target B. subtilis strain, CRISPR plasmid, E. coli dam-/dcm- methyltransferase-deficient strain, in vitro CpG methyltransferase (M.SssI), glycine. Procedure:
- Methylated Plasmid Preparation: Isolate plasmid from a standard E. coli strain (e.g., DH5α). Treat 2 µg of plasmid with M.SssI methyltransferase per manufacturer's protocol (37°C, 1 hour). Purify using a standard PCR clean-up kit. Alternatively, propagate plasmid in a dam-/dcm- E. coli strain before M.SssI treatment.
- Culture Pre-treatment: Grow target B. subtilis in LB supplemented with 0.5-1% (w/v) glycine to mid-log phase. Glycine weakens peptidoglycan cross-linking.
- Competent Cell Preparation: Use standard chemical competence protocol (e.g., CaCl2 method) or electrocompetent cells prepared from glycine-treated culture.
- Transformation: For chemical transformation, mix 100 µL competent cells with 100 ng methylated plasmid. Heat shock at 37°C for 30 minutes (for Bacillus). Add recovery medium, incubate with shaking for 90 minutes, then plate.
Visualization: Workflows and Pathways
Title: Decision Workflow for Overcoming CRISPR Barriers
Title: Toxicity Pathway vs. Mitigation Strategy
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Reagent / Material | Function & Application in Refractory Organisms |
|---|---|
| Chemically Modified sgRNA (2'-O-methyl, phosphorothioate) | Increases nuclease resistance and RNP stability during delivery, crucial for harsh cellular environments. |
| High-Purity Cas9 Protein (WT or HiFi) | Essential for RNP approaches. Reduces off-target effects (HiFi variant) and host transcriptional burden. |
| Host-Specific or Inducible Expression Vector | Plasmid with a native origin of replication (ori) and a tightly regulated promoter (e.g., xylose, tetracycline) to control Cas9 timing. |
| CpG Methyltransferase (M.SssI) | In vitro methylation of plasmid DNA to protect against restriction enzyme degradation in wild-type strains. |
| Cell Wall Weakening Agents (e.g., Glycine, D-Cycloserine) | Pre-treatment additives to inhibit peptidoglycan synthesis, increasing permeability for macromolecules. |
| Electroporation Enhancers (e.g., Sucrose, Glycerol) | Osmoprotectants in electroporation buffers to increase cell survival after electrical pulse. |
| dam-/dcm- E. coli Strains | For producing plasmid DNA lacking common methylation patterns that trigger restriction systems. |
| Cas9 "Kill Switch" Plasmid | A self-deleting vector system that expresses Cas9/sgRNA then removes itself via recombinase, minimizing persistent toxicity. |
Within metabolic engineering research utilizing CRISPR/Cas9, precise genomic modifications are paramount for redirecting metabolic fluxes or introducing novel biosynthetic pathways. The efficacy of Cas9-mediated editing is fundamentally governed by the design of the single-guide RNA (sgRNA). Computational tools and algorithmic predictors are indispensable for identifying sgRNAs with high on-target activity and minimal off-target effects, thereby accelerating the engineering of microbial or mammalian cell factories for therapeutic compound production.
Computational Predictors for On-Target Efficiency
Modern algorithms integrate multiple sequence and structural features to predict sgRNA cutting efficiency. Key features include GC content, specific nucleotide preferences at certain positions, melting temperature, and chromatin accessibility data. The following table summarizes leading tools and their core predictive features.
Table 1: Comparison of Key sgRNA On-Target Efficiency Predictors
| Tool Name | Key Predictive Features | Model Basis | Access |
|---|---|---|---|
| DeepSpCas9 | Sequence context, DNA duplex stability, chromatin accessibility (if provided) | Deep neural network trained on large-scale libraries | Web server, Standalone |
| CRISPOR | Multiple scoring algorithms (Doench '16, Moreno-Mateos, etc.), off-target analysis | Rule-based and regression models integrating published data | Web server, Command line |
| Rule Set 2 | 30-nt sequence context, GC content, specific position nucleotides | Regularized linear regression model | Built into IDT's design tool |
| CRISPRscan | Sequence features, nucleotide composition, zebrafish embryo data | Gradient boosting machine learning | Web server |
Protocols for In Silico sgRNA Design and Evaluation
Protocol 3.1: Comprehensive sgRNA Selection for a Metabolic Engineering Target
Objective: To design high-efficiency sgRNAs targeting a gene (e.g., fass in yeast) for knockout to enhance precursor flux toward a desired product. Materials: Computer with internet access; target gene sequence (FASTA format). Procedure:
- Sequence Retrieval: Obtain the coding sequence (CDS) of your target gene from a reliable database (e.g., NCBI, Ensembl).
- Candidate Identification: Use a basic scanner (e.g., CRISPRdirect, E-CRISP) to identify all protospacer adjacent motif (PAM) sequences (NGG for SpCas9) and extract the 20-nt upstream sequences as candidate sgRNAs.
- On-Target Scoring: Input the list of candidate sgRNAs into at least two predictors from Table 1 (e.g., CRISPOR and DeepSpCas9). Rank candidates by their predicted efficiency scores.
- Off-Target Assessment: For the top 5-10 candidates, perform a genome-wide off-target search using tools integrated in CRISPOR or Cas-OFFinder. Set parameters: allow up to 3 mismatches. Discard any sgRNA with a perfect or near-perfect match (≤2 mismatches) elsewhere in the genome.
- Final Selection: Prioritize sgRNAs with high on-target scores (>70th percentile), no predicted high-risk off-targets, and targeting an early exon for protein null alleles.
Protocol 3.2: Experimental Validation of Predicted Efficiency (T7E1 Assay)
Objective: To empirically validate the cleavage efficiency of computationally selected sgRNAs. Materials: Designed sgRNA constructs or synthetic sgRNAs; Cas9 expression vector; target cell line; PCR reagents; T7 Endonuclease I (NEB); agarose gel electrophoresis system. Procedure:
- Transfection: Co-transfect your target cells with the Cas9 plasmid and each candidate sgRNA expression plasmid (or ribonucleoprotein complexes). Include a negative control (Cas9 only).
- Genomic DNA Extraction: 72 hours post-transfection, harvest cells and extract genomic DNA.
- PCR Amplification: Design primers flanking the target site (~500-800 bp amplicon). PCR-amplify the target region from all samples.
- Heteroduplex Formation: Denature and reanneal PCR products: 95°C for 5 min, ramp down to 85°C at -2°C/sec, then to 25°C at -0.1°C/sec.
- T7E1 Digestion: Digest reannealed products with T7 Endonuclease I for 30 min at 37°C. This enzyme cleaves heteroduplex DNA formed by wild-type and edited strands.
- Analysis: Run digested products on an agarose gel. Quantify cleavage efficiency using band intensities: % indel = 100 * (1 - sqrt(1 - (b+c)/(a+b+c))), where a is the intact band, and b & c are cleavage products.
Diagrams
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for sgRNA Design and Validation Experiments
| Item | Function / Explanation |
|---|---|
| Synthetic sgRNA or Cloning Kit | For rapid sgRNA delivery; synthetic sgRNAs allow immediate RNP formation, while kits (e.g., Addgene's CRISPR plasmids) enable stable expression. |
| High-Fidelity Cas9 Expression Vector | Ensures precise and efficient DNA cleavage. Nuclease-dead (dCas9) variants are used for transcriptional control in metabolic engineering. |
| Genomic DNA Extraction Kit | To obtain high-quality template DNA from edited cells for downstream validation assays (PCR, sequencing). |
| T7 Endonuclease I | Enzyme for the mismatch cleavage assay, a cost-effective method for initial indel detection and efficiency estimation. |
| Next-Generation Sequencing (NGS) Library Prep Kit | For deep sequencing of the target locus, providing the most accurate quantification of editing efficiency and spectrum of indels. |
| Cell Line-Specific Transfection Reagent | Critical for efficient delivery of CRISPR components into the host cell (e.g., lipofection, electroporation reagents). |
| Homology-Directed Repair (HDR) Donor Template | Single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA containing the desired edit for precise metabolic pathway engineering. |
Within metabolic engineering research, precision genome editing using CRISPR/Cas9 enables targeted optimization of enzymatic pathways, knockout of competing reactions, and insertion of heterologous genes. The selection of the appropriate Cas9 variant is critical, balancing editing efficiency, specificity, delivery constraints, and target site availability. This application note provides a comparative analysis of commonly used Cas9 variants and detailed protocols for their application in metabolic pathway engineering.
Comparative Analysis of Cas9 Variants
Table 1: Key Characteristics of Primary Cas9 Variants
| Variant | PAM Sequence | Size (aa) | Key Strengths | Primary Limitations | Ideal Use Case in Metabolic Engineering |
|---|---|---|---|---|---|
| spCas9 (Streptococcus pyogenes) | 5'-NGG-3' | 1368 | High efficiency; extensive validation; wide reagent availability. | Large size; off-target effects; PAM restriction. | High-efficiency editing in easily transfected cells (e.g., yeast, CHO, HEK293) for pathway knockout. |
| saCas9 (Staphylococcus aureus) | 5'-NNGRRT-3' | 1053 | Smaller size; good efficiency; alternative PAM. | Lower efficiency than spCas9 in some contexts; some off-target risk. | AAV-delivery for in vivo or primary cell editing; expands targetable genomic sites. |
| spCas9-HF1 | 5'-NGG-3' | 1368 | Dramatically reduced off-target cleavage. | Can have reduced on-target efficiency. | Editing essential genes where off-targets could disrupt cell metabolism. |
| eSpCas9(1.1) | 5'-NGG-3' | 1368 | Reduced off-target activity. | Can have reduced on-target efficiency. | Multi-locus editing to balance multiple pathway enzymes without genotoxic stress. |
| evoCas9 | 5'-NGG-3' | 1368 | High-fidelity, maintained efficiency. | Proprietary; may require optimization. | Engineering producer cell lines for biotherapeutics where clonal purity is paramount. |
Table 2: Performance Metrics in Mammalian Cells (Representative Data)
| Variant | On-Target Indel Efficiency (%)* | Relative Off-Target Activity* | Reference |
|---|---|---|---|
| Wild-type spCas9 | 40-80 | 1.0 (Baseline) | Cong et al., 2013 |
| saCas9 | 30-60 | ~0.8-1.2 | Ran et al., 2015 |
| spCas9-HF1 | 20-60 | <0.01 | Kleinstiver et al., 2016 |
| eSpCas9(1.1) | 25-65 | ~0.02 | Slaymaker et al., 2016 |
| evoCas9 | 40-75 | <0.01 | Casini et al., 2018 |
*Ranges are context-dependent. Off-target activity measured at known problematic sites.
Application Notes for Metabolic Engineering
Variant Selection Workflow
The choice of Cas9 variant should follow a decision tree based on experimental priorities: delivery method, target sequence availability, and required fidelity.
Title: Cas9 Variant Selection Decision Tree
Key Signaling Pathway: DNA Damage Response in Edited Cells
Understanding cellular response to double-strand breaks (DSBs) is crucial for designing editing strategies, especially when making multiple edits to a metabolic network.
Title: DNA Damage Repair Pathways After Cas9 Cleavage
Detailed Experimental Protocols
Protocol 1: Multiplexed Gene Knockout in Yeast for Pathway Engineering Using spCas9
Objective: Simultaneously knock out 2-3 genes encoding enzymes in a competing metabolic branch to flux carbon toward a desired product.
Materials (The Scientist's Toolkit):
| Reagent/Material | Function/Description |
|---|---|
| pCAS-yeast (spCas9 expression) | Plasmid expressing spCas9 and a selectable marker for the host yeast. |
| gRNA Expression Plasmid(s) | Contains tandem gRNA expression cassettes (e.g., using tRNA processing system). |
| Homology Repair Template(s) | Optional, short oligonucleotides for precise edits or to introduce a stop codon. |
| YPAD or Selective Media | For yeast cultivation and plasmid maintenance. |
| LiAc/SS Carrier DNA/PEG Solution | Components for standard yeast transformation (LiAc method). |
| Cas9 Nuclease Assay Kit | Optional, for verifying Cas9 activity in cell lysates. |
| Surveyor/Nuclease or T7E1 | For initial validation of editing efficiency at the bulk population level. |
| PCR Reagents & Sanger Sequencing Primers | For amplification and sequencing of target loci from clones. |
Procedure:
- Design: For each target gene, design a gRNA targeting an early exon. Ensure spCas9 PAM (NGG) is present. Design PCR primers ~300-500 bp flanking each cut site.
- Cloning: Clone individual gRNA sequences into a multiplex-compatible yeast gRNA vector. Co-transform this vector and the pCAS-yeast plasmid into your yeast strain using the high-efficiency LiAc method. Plate on appropriate double-selection media.
- Screening (Bulk): After 2-3 days growth, harvest a portion of the transformation pool. Extract genomic DNA. PCR-amplify each target region. Run T7 Endonuclease I (T7E1) assay on the purified PCR products to assess bulk editing efficiency.
- Isolation of Clones: Dilute and spread the transformation on fresh plates to obtain single colonies. Pick 20-50 colonies and patch/re-streak.
- Genotyping: Perform colony PCR on each target locus for each clone. Send PCR products for Sanger sequencing. Analyze chromatograms for indels using tools like ICE (Inference of CRISPR Edits) or TIDE.
- Validation: For confirmed multiplex knockout clones, conduct phenotyping (e.g., growth assay in relevant substrate) and metabolomic analysis (e.g., HPLC/MS) to verify redirected metabolic flux.
Protocol 2: High-Fidelity Editing of a Metabolic Enzyme Gene in Mammalian Cells Using evoCas9
Objective: Introduce a precise, single amino acid substitution (knock-in) in a gene encoding a rate-limiting enzyme without off-target mutations that could confound phenotyping.
Materials (The Scientist's Toolkit):
| Reagent/Material | Function/Description |
|---|---|
| evoCas9 Expression Plasmid | Plasmid encoding the high-fidelity evoCas9 nuclease. |
| gRNA Expression Construct | U6-driven gRNA expression vector. |
| Single-Stranded Oligodeoxynucleotide (ssODN) | ~100-200 nt HDR template containing the desired point mutation and synonymous PAM-disrupting changes. |
| HEK293T or Relevant Cell Line | Model mammalian cells. |
| Lipofectamine 3000 or Electroporation System | Transfection reagents. |
| Genomic DNA Extraction Kit | For harvesting DNA from edited pools and clones. |
| Next-Generation Sequencing (NGS) Library Prep Kit | For comprehensive on-target and off-target analysis. |
| Cloning Medium & FACS/Limiting Dilution Supplies | For isolation of single-cell clones. |
Procedure:
- Design: Design gRNA with an NGG PAM as close to the target codon as possible. Design the ssODN homology-directed repair (HDR) template with the desired edit, flanked by 50-80 nt homology arms. Include silent mutations in the PAM or seed sequence to prevent re-cutting.
- Transfection: Seed cells in a 24-well plate. Co-transfect 500 ng evoCas9 plasmid, 250 ng gRNA plasmid, and 100-200 pmol of ssODN using Lipofectamine 3000 per manufacturer's protocol.
- Initial Assessment: At 72 hours post-transfection, extract genomic DNA from the pool. Amplify the target region by PCR and sequence via Sanger or NGS to estimate HDR efficiency.
- Single-Cell Cloning: At 48h post-transfection, trypsinize and dilute cells to ~0.5 cells/well in a 96-well plate. Culture for 2-3 weeks.
- Clone Screening: Expand clones, extract gDNA, and PCR-amply the target region. Perform Sanger sequencing to identify heterozygous and homozygous HDR clones.
- Off-Target Validation: Identify top 5-10 potential off-target sites using an in-silico predictor (e.g., Cas-OFFinder). Perform PCR and deep sequencing (amplicon-seq) on these loci from the positive clone and a wild-type control. Confirm absence of significant indel formation above background sequencing error.
- Functional Assay: Measure enzyme activity (e.g., via spectrophotometric assay) and overall metabolic output (e.g., product titer) in the engineered clone versus parental control.
Workflow: From Design to Validation
Title: End-to-End CRISPR-Cas9 Metabolic Engineering Workflow
For metabolic engineering, spCas9 remains the workhorse for standard, high-efficiency knockouts. saCas9 is vital for delivery-constrained contexts. When engineering complex traits where genetic purity is essential, such as creating industrial producer cell lines, high-fidelity mutants like evoCas9, spCas9-HF1, or eSpCas9(1.1) are indispensable to avoid confounding off-target metabolic effects. The protocol selected must align with the variant's strengths to precisely rewire cellular metabolism.
Validating Edits and Benchmarking CRISPR/Cas9 Against Traditional Metabolic Engineering Tools
Application Notes
In the context of a thesis on CRISPR/Cas9 for metabolic engineering, rigorous validation of edits is paramount. These techniques confirm on-target modifications, detect off-target effects, and verify functional phenotypic outcomes, ensuring engineered microbial or cell line models accurately reflect the desired metabolic pathway alterations.
Sanger Sequencing remains the gold standard for validating specific, targeted edits. It provides high accuracy for confirming point mutations, small insertions/deletions (indels), and short homology-directed repair (HDR) events at defined loci. Its application is critical for final clone verification following single-cell isolation.
Next-Generation Sequencing (NGS) enables comprehensive validation. Amplicon-based deep sequencing quantitatively assesses editing efficiency and identifies allelic heterogeneity at target sites. Whole-genome or exome sequencing is essential for unbiased genome-wide off-target profiling, a crucial step for therapeutic and industrial strain development.
Phenotypic Screening validates functional consequences. For metabolic engineering, this includes assays for metabolite production (e.g., via HPLC/MS), growth under selective conditions, or fluorescence-based reporters for pathway activation. It links genotypic changes to the desired physiological output.
Table 1: Comparative Summary of Validation Techniques
| Technique | Primary Application in CRISPR Validation | Key Metric | Approximate Cost per Sample (USD) | Time to Result |
|---|---|---|---|---|
| Sanger Sequencing | Confirmation of intended edits at a specific locus. | Sequence chromatogram quality, base call accuracy. | $10 - $20 | 1-2 days |
| NGS (Amplicon-Seq) | Quantifying editing efficiency & analyzing mutation spectra. | Read depth (≥1000x), variant allele frequency (%). | $50 - $200 | 3-7 days |
| NGS (WGS) | Genome-wide off-target effect discovery. | Coverage (≥30x), off-target site identification. | $1000 - $3000 | 1-2 weeks |
| Phenotypic Screening | Assessing functional impact of edits. | Metabolite titer, growth rate, fluorescence intensity. | Variable ($20 - $500) | 1 day - 1 week |
Detailed Protocols
Protocol 1: Sanger Sequencing for CRISPR Edit Verification
Objective: To confirm the precise DNA sequence at the CRISPR/Cas9-targeted genomic locus in cloned engineered cells.
Materials:
- Purified genomic DNA from clonal population.
- Target-specific PCR primers (flanking the edit site).
- PCR master mix, agarose gel electrophoresis equipment.
- PCR purification kit, sequencing primer.
- Capillary sequencer access.
Procedure:
- Amplify Target Locus: Perform PCR (35 cycles) using gene-specific primers to generate a 300-800 bp amplicon encompassing the edited region.
- Purify Amplicon: Resolve PCR product on agarose gel, excise correct band, and purify using a gel extraction kit. Quantify DNA.
- Prepare Sequencing Reaction: In a 10 µL reaction, mix 1-10 ng of purified amplicon, 5 pmol of sequencing primer (one of the PCR primers or an internal primer), and sequencing mix (BigDye Terminator v3.1).
- Cycle Sequencing: Run: 96°C for 1 min, then 25 cycles of [96°C for 10 sec, 50°C for 5 sec, 60°C for 4 min].
- Purification & Sequencing: Purify reaction using a column or ethanol precipitation. Run on capillary sequencer.
- Analysis: Align sequencing chromatogram to reference sequence using software (e.g., SnapGene, Sequencher) to identify insertions, deletions, or substitutions.
Protocol 2: NGS Amplicon Sequencing for Editing Efficiency
Objective: To quantify the spectrum and frequency of indel mutations at the on-target site.
Materials:
- Genomic DNA from polyclonal or clonal population.
- High-fidelity DNA polymerase (e.g., Q5, KAPA HiFi).
- Primers with overhang adapters for index attachment.
- Library preparation kit (e.g., Illumina Nextera XT).
- NGS platform (e.g., Illumina MiSeq).
Procedure:
- Primary PCR (Target Amplification): Design primers ~150-200 bp flanking cut site. Perform PCR with high-fidelity polymerase to generate initial amplicon. Clean up product.
- Indexing PCR (Library Construction): Add universal Illumina adapter sequences and unique dual indices via a limited-cycle (8-10 cycles) PCR. Pool barcoded samples.
- Library Purification & Quantification: Clean pooled library with SPRI beads. Precisely quantify using qPCR (KAPA Library Quant Kit).
- Sequencing: Dilute library to 4 nM, denature, and load onto MiSeq cartridge for 2x250 bp or 2x300 bp paired-end sequencing to ensure overlap.
- Bioinformatics Analysis:
- Demultiplex reads by sample indices.
- Merge paired-end reads (FLASH, PEAR).
- Align merged reads to reference amplicon sequence (BWA, CRISPResso2).
- Quantify percent of reads with indels and characterize indel size distribution.
Protocol 3: Phenotypic Screening via Metabolite Titer Analysis (HPLC)
Objective: To validate enhanced production of a target metabolite (e.g., succinate) in CRISPR-engineered yeast strains.
Materials:
- Engineered and wild-type control strains in appropriate culture medium.
- Microplate reader or spectrophotometer.
- HPLC system with UV/RI or MS detector.
- Appropriate analytical column (e.g., Aminex HPX-87H for organic acids).
- Metabolite standard for quantification.
Procedure:
- Culture Growth: Inoculate engineered and control strains in triplicate in minimal medium with limiting nitrogen to trigger metabolite production. Grow in deep-well plates at 30°C, 300 rpm for 48-72 hrs.
- Sample Preparation: Take 1 mL aliquots at stationary phase. Centrifuge at 13,000 x g for 5 min to pellet cells. Filter supernatant through 0.2 µm syringe filter.
- HPLC Analysis:
- Column: Aminex HPX-87H, 300 x 7.8 mm.
- Mobile Phase: 5 mM H₂SO₄, isocratic flow of 0.6 mL/min.
- Column Temp: 45°C.
- Detection: Refractive Index (RI) detector.
- Run Time: 30 min.
- Quantification: Inject 10 µL of filtered supernatant. Identify succinate peak by retention time matching to standard curve (e.g., 0.1, 0.5, 1, 5, 10 g/L). Integrate peak areas.
- Data Analysis: Compare average succinate titer (g/L) between engineered and control strains using a Student's t-test. A statistically significant increase (p < 0.05) confirms functional phenotypic impact.
Diagrams
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CRISPR Validation
| Item | Function in Validation | Example Product/Brand |
|---|---|---|
| High-Fidelity DNA Polymerase | Reduces PCR errors during amplicon generation for sequencing. | Q5 (NEB), KAPA HiFi (Roche) |
| PCR Clean-Up & Gel Extraction Kits | Purifies DNA fragments to remove primers, enzymes, and salts for downstream applications. | NucleoSpin Gel and PCR Clean-up (Macherey-Nagel) |
| Sanger Sequencing Reagents | Fluorescent dye-terminator chemistry for capillary sequencing. | BigDye Terminator v3.1 (Thermo Fisher) |
| NGS Library Prep Kit | Attaches sequencing adapters and sample indices for multiplexing. | Nextera XT DNA Library Prep Kit (Illumina) |
| Library Quantification Kit | Accurate qPCR-based quantification of NGS libraries for optimal loading. | KAPA Library Quantification Kit (Roche) |
| CRISPR Analysis Software | Bioinformatics tool for quantifying editing from NGS data. | CRISPResso2, ICE (Synthego) |
| Metabolite Standards | Pure chemical for generating calibration curves in phenotypic HPLC/MS. | Succinic Acid, Sigma-Aldrich |
| HPLC Column | Separates metabolites in complex culture supernatants for quantification. | Aminex HPX-87H Ion Exclusion Column (Bio-Rad) |
Metabolic Flux Analysis (MFA) to Quantify Pathway Rewiring and Product Yield
Within a thesis on CRISPR/Cas9 genome editing for metabolic engineering, Metabolic Flux Analysis (MFA) serves as the critical, quantitative framework for assessing the functional consequences of genetic perturbations. Following genome editing, it is insufficient to merely confirm gene knockout or insertion; one must quantify how carbon flow is redirected through metabolic networks. This application note details how (^{13})C-based MFA protocols are employed post-CRISPR editing to rigorously quantify pathway rewiring and product yield enhancement in microbial or cell culture systems, providing the data necessary to iteratively guide engineering strategies.
Core Principles & Application in CRISPR Engineering
MFA calculates the in vivo flow of metabolites through biochemical reactions in a metabolic network at isotopic steady state. In the context of CRISPR engineering:
- Pre-Edit Baseline: (^{13})C-MFA establishes a flux map for the wild-type or parent strain.
- Post-Edit Validation: After CRISPR-mediated knockout (e.g., of a competing pathway gene) or activation (e.g., of a rate-limiting enzyme), (^{13})C-MFA quantifies the resultant flux redistribution.
- Yield Analysis: The net flux toward the target product (e.g., succinate, taxadiene, a therapeutic antibody) is directly calculated, providing an absolute measure of engineering success beyond extracellular titers.
Key Quantitative Outputs:
- Flux redistribution at key metabolic branch points (e.g., glycolytic split, TCA node).
- Change in cofactor (NADPH, ATP) generation/consumption.
- Absolute product yield (mol product / mol substrate).
Experimental Protocols
Protocol 3.1: Tracer Experiment for (^{13})C-MFA Post-CRISPR Editing
Objective: To introduce a (^{13})C-labeled substrate into CRISPR-edited and control cultures for subsequent flux analysis.
Materials: CRISPR-edited strain, isogenic control strain, chemically defined medium, (^{13})C-labeled substrate (e.g., [1-(^{13})C]glucose, [U-(^{13})C]glucose), bioreactor or controlled shake flasks, filtration/sampling setup.
Procedure:
- Pre-culture: Grow both control and edited strains in unlabeled medium to mid-exponential phase.
- Wash & Transfer: Harvest cells via centrifugation, wash twice with PBS or medium base, and inoculate into fresh medium containing the (^{13})C-labeled substrate at the same concentration as standard medium. Ensure optical density (OD) is sufficient for metabolic steady-state.
- Steady-State Cultivation: Maintain cultures in a bioreactor or controlled environment (constant pH, temperature, dissolved O₂) for a duration exceeding 5-6 generation times to ensure isotopic steady state is reached.
- Metabolite Quenching & Sampling:
- Rapidly sample culture (e.g., into cold 60% methanol quenching solution) for intracellular metabolite analysis.
- Simultaneously, sample extracellular medium for substrate consumption and product secretion rates (HPLC/GC analysis).
- Harvest cell pellet for biomass composition analysis (protein, glycogen, lipids) and dry cell weight determination.
Protocol 3.2: GC-MS Sample Preparation & Measurement for (^{13})C Labeling
Objective: To derive mass isotopomer distribution vectors (MIDs) of proteinogenic amino acids from cellular biomass.
Materials: Harvested cell pellet, 6M HCl, nitrogen evaporation system, derivatization agents [N-methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide (MTBSTFA) + 1% tert-butyldimethylchlorosilane (TBDMCS) or N,N-Dimethylformamide dimethyl acetal (DMF-DMA)], GC-MS system.
Procedure:
- Hydrolysis: Hydrolyze dried biomass pellet in 6M HCl at 105°C for 24 hours to release free amino acids.
- Drying: Dry the hydrolysate under a stream of nitrogen to remove HCl.
- Derivatization:
- For MTBSTFA: Reconstitute in pyridine, add derivatization agent, incubate at 85°C for 1 hour.
- For DMF-DMA (for GC-TOF): Use a dedicated autosampler protocol.
- GC-MS Analysis: Inject sample. Use a standard non-polar column (e.g., DB-5MS). Method: hold at 100°C, ramp to 300°C. Operate MS in electron impact (EI) mode.
- Data Processing: Integrate chromatogram peaks. For each amino acid fragment (e.g., alanine [m-57]⁺), calculate the fractional abundance of mass isotopomers M₀, M₁, M₂,... Mₙ to form the MID.
Protocol 3.3: Computational Flux Estimation
Objective: To calculate metabolic fluxes from experimental MIDs and extracellular rates.
Materials: Measured MIDs, substrate uptake/product secretion rates, biomass composition, stoichiometric metabolic network model (e.g., for E. coli, S. cerevisiae, CHO cells), software (INCA, 13C-FLUX2, OpenFLUX).
Procedure:
- Network Definition: Construct or load a stoichiometrically balanced model including central carbon metabolism (glycolysis, PPP, TCA, etc.).
- Data Input: Input measured extracellular fluxes and labeling data (MIDs).
- Flux Estimation: Use software to perform an iterative least-squares regression, minimizing the difference between simulated and measured MIDs. The algorithm adjusts net and exchange fluxes within the network.
- Statistical Evaluation: Software provides estimated fluxes with confidence intervals (from Monte Carlo or sensitivity analysis). Fluxes with confidence intervals not spanning zero are considered statistically significant.
Data Presentation
Table 1: Comparative Flux Analysis at Key Nodes Pre- and Post-CRISPR Knockout Example: Knockout of *ldhA in E. coli for succinate production.*
| Metabolic Reaction / Branch Point | Flux in Control Strain (mmol/gDCW/h) | Flux in CRISPR-Edited Strain (mmol/gDCW/h) | % Change | P-value |
|---|---|---|---|---|
| Glucose Uptake | 10.0 ± 0.5 | 9.8 ± 0.6 | -2.0 | 0.65 |
| Glycolysis (to G3P) | 8.5 ± 0.4 | 9.2 ± 0.5 | +8.2 | 0.04 |
| Pentose Phosphate Pathway | 1.5 ± 0.2 | 1.8 ± 0.3 | +20.0 | 0.12 |
| Pyruvate Kinase (to Pyruvate) | 7.0 ± 0.4 | 8.5 ± 0.5 | +21.4 | <0.01 |
| Lactate Dehydrogenase (LDH) | 5.2 ± 0.3 | 0.1 ± 0.05 | -98.1 | <0.001 |
| Pyruvate Dehydrogenase (to Acetyl-CoA) | 1.5 ± 0.2 | 2.1 ± 0.3 | +40.0 | 0.03 |
| Anaerobic Succinate Pathway | 0.8 ± 0.1 | 4.7 ± 0.4 | +487.5 | <0.001 |
| TCA Cycle (Oxaloacetate Turnover) | 2.0 ± 0.3 | 1.5 ± 0.2 | -25.0 | 0.05 |
Table 2: Product Yield Metrics Before and After Pathway Rewiring
| Metric | Control Strain | CRISPR-Edited Strain | Improvement Factor |
|---|---|---|---|
| Succinate Yield (mol/mol Glc) | 0.08 ± 0.01 | 0.48 ± 0.04 | 6.0x |
| Max Theoretical Yield (%) | 15% | 90% | |
| Biomass Yield (gDCW/mol Glc) | 28.5 ± 1.2 | 22.1 ± 1.5 | - |
| Redox Cofactor Balance (NADH/NAD⁺) | 1.05 ± 0.05 | 0.91 ± 0.06 | More Oxidized |
Visualizations
Title: Central Carbon Flux Map in Control Strain
Title: Flux Rewiring After CRISPR Knockout of LDH
Title: 13C-MFA Experimental & Computational Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for 13C-MFA in Metabolic Engineering
| Item / Reagent | Function in Protocol | Key Consideration |
|---|---|---|
| Uniformly Labeled [U-13C] Glucose | Primary tracer substrate for comprehensive labeling of central carbon metabolites. | Ensures high information content for flux resolution; isotopic purity >99%. |
| Positionally Labeled Tracers (e.g., [1-13C] Glc) | Used for specific pathway resolution (e.g., PPP vs. glycolysis). | Selected based on network topology and specific flux questions. |
| MTBSTFA + 1% TBDMCS | Derivatization agent for GC-MS analysis of amino acids. Produces stable tert-butyldimethylsilyl (TBDMS) derivatives. | Must be handled under anhydrous conditions; hygroscopic. |
| DMF-DMA (N,N-Dimethylformamide dimethyl acetal) | Derivatization agent for GC-TOF MS, creating methyl esters. | Faster reaction, suitable for high-throughput automation. |
| Stable Isotope-Labeled Amino Acid Standards | Internal standards for LC-MS based MFA or calibration. | Correct for instrument variability and ionization efficiency. |
| INCA Software (or 13C-FLUX2, OpenFLUX) | Platform for metabolic network modeling, flux simulation, and statistical fitting of 13C labeling data. | Requires a correctly curated stoichiometric model of the organism. |
| Chemically Defined Medium | Essential for precise control of substrate concentration and absence of unlabeled carbon sources. | Eliminates background labeling noise from complex nutrients like yeast extract. |
Within metabolic engineering research, the primary goal is to rewire cellular metabolism to produce high-value compounds. This requires precise, stable genetic modifications to upregulate, downregulate, or knock out specific metabolic pathway genes. CRISPR/Cas9, Homologous Recombination (HR), and RNA Interference (RNAi) represent three pivotal technologies for achieving these goals, each with distinct mechanisms, applications, and limitations.
Core Mechanisms
- CRISPR/Cas9: A bacterial adaptive immune system repurposed for genome editing. The Cas9 nuclease is guided by a single-guide RNA (sgRNA) to a specific genomic locus, where it creates a double-strand break (DSB). This break is repaired by non-homologous end joining (NHEJ), often causing insertions/deletions (indels) and gene disruption, or by homology-directed repair (HDR) using a donor DNA template for precise edits.
- Homologous Recombination (HR): A natural cellular process for repairing DSBs. In classical gene targeting, a designed donor vector with long homology arms (>>1 kb) recombines with the target genomic locus at low frequency, enabling precise gene knock-in, knockout, or replacement. Highly efficient in mouse embryonic stem cells but inefficient in most other cell types without additional tools.
- RNA Interference (RNAi): A post-transcriptional gene silencing mechanism. Double-stranded RNA (dsRNA) is processed by Dicer into small interfering RNAs (siRNAs). These are loaded into the RNA-induced silencing complex (RISC), which binds and cleaves complementary messenger RNA (mRNA), leading to degradation and transient knockdown of gene expression without altering the DNA sequence.
Quantitative Comparison Table
Table 1: Head-to-Head Technical Comparison
| Feature | CRISPR/Cas9 (with HDR) | Homologous Recombination (Classical) | RNAi (siRNA/shRNA) |
|---|---|---|---|
| Primary Use | Gene knockout, precise knock-in, repression/activation (via dCas9) | Precise gene knockout, knock-in, or replacement | Transient gene knockdown |
| Target | Genomic DNA | Genomic DNA | mRNA (cytoplasm) |
| Edit Precision | Very High (with HDR template) | Very High | N/A (no genomic change) |
| Permanence | Stable, heritable | Stable, heritable | Transient (days to weeks) |
| Efficiency | High to Very High (NHEJ); Moderate (HDR) | Very Low (in most somatic cells) | High (knockdown >70% common) |
| Multiplexing | High (multiple sgRNAs) | Very Low | Moderate (multiple siRNAs) |
| Off-Target Effects | Moderate (DNA-level; improved with high-fidelity Cas9) | Very Low | High (RNA-level; seed region matches) |
| Delivery | Plasmid, ribonucleoprotein (RNP) | Large targeting vectors, often requiring selection | siRNA (transfection), shRNA (viral) |
| Throughput | High (pooled libraries) | Low | High (arrayed screens) |
| Key Challenge | Optimizing HDR efficiency; off-target edits | Extremely low efficiency in primary cells | Transient effect; compensatory responses |
Table 2: Application Suitability for Metabolic Engineering
| Application Goal | Recommended Technology | Rationale |
|---|---|---|
| Complete gene knockout | CRISPR/Cas9 (NHEJ) | High efficiency, stable, enables multiplexing of pathway enzymes. |
| Precise point mutation | CRISPR/Cas9 (HDR) | Allows single-base changes in enzymes or regulators with donor template. |
| Large DNA insertions | CRISPR/Cas9 or HR (if in ES cells) | CRISPR HDR works in many systems; HR remains gold standard for large inserts in mouse ES cells. |
| Rapid gene knockdown screen | RNAi | Fast, well-established libraries for identifying metabolic pathway bottlenecks. |
| Tuning gene expression | CRISPRi/a (dCas9) | Enables stable, programmable repression (CRISPRi) or activation (CRISPRa) of promoters. |
| Engineering primary cells | CRISPR/Cas9 RNP | High efficiency, reduced off-targets and toxicity compared to plasmid delivery. |
Detailed Application Notes & Protocols
Protocol: CRISPR/Cas9-Mediated Gene Knockout in Mammalian Cells for Metabolic Flux Analysis
Aim: To disrupt a key regulatory gene (e.g., PDK1) in HEK293 cells to shift flux from glycolysis to oxidative phosphorylation.
Materials (The Scientist's Toolkit):
- HEK293T Cells: Common mammalian cell line with high transfection efficiency.
- Lipofectamine CRISPRMAX: Lipid-based transfection reagent optimized for RNP delivery.
- Alt-R S.p. HiFi Cas9 Nuclease V3: High-fidelity Cas9 protein for reduced off-target effects.
- Alt-R CRISPR-Cas9 sgRNA: Chemically synthesized, modified sgRNA for high stability and efficiency.
- Alt-R HDR Enhancer V2: Small molecule to improve HDR efficiency if performing knock-in.
- Cell Culture Media (DMEM + 10% FBS): Standard growth medium.
- Genomic DNA Purification Kit: For isolating DNA for genotyping.
- T7 Endonuclease I or ICE Analysis Tool: For detecting indel mutations.
- Flow Cytometer or Fluorescence Microscope: For analyzing fluorescent reporters if used.
Procedure:
- sgRNA Design: Use a validated online tool (e.g., CHOPCHOP, CRISPOR) to design sgRNAs targeting early exons of the PDK1 gene. Select top-ranked guides with minimal predicted off-targets.
- RNP Complex Formation: For one well of a 24-well plate, combine 3 µl of 10 µM sgRNA with 1.5 µl of 10 µM Alt-R Cas9 HiFi protein. Incubate at room temperature for 10-20 minutes.
- Cell Transfection: Seed 1.5 x 10^5 HEK293T cells/well. Prepare lipid complex: Dilute 1.5 µl CRISPRMAX in 25 µl Opti-MEM. Mix with RNP complex, incubate 10 min, then add dropwise to cells.
- Analysis & Cloning: 72 hours post-transfection, harvest cells. Extract genomic DNA. PCR-amplify the target region. Use T7E1 assay or Sanger sequencing followed by ICE analysis to quantify editing efficiency.
- Single-Cell Cloning: If a pure knockout population is needed, perform serial dilution to isolate single cells. Expand clonal lines and screen by sequencing to identify homozygous knockouts.
- Metabolic Phenotyping: Validate knockout by western blot (loss of protein). Measure metabolic flux via Seahorse Analyzer (increased OCR, decreased ECAR).
Protocol: RNAi-Mediated Knockdown for Rapid Identification of Metabolic Limiting Steps
Aim: To screen a panel of siRNAs targeting enzymes in a heterologous biosynthesis pathway to identify yield-limiting steps.
Procedure:
- siRNA Library Design: Obtain a validated siRNA library (e.g., Silencer Select) targeting 5-10 genes in your pathway of interest. Include non-targeting (scramble) and positive control (essential gene) siRNAs.
- Reverse Transfection in 96-well Plate: Using a lipid transfection reagent, complex 10 nM of each siRNA with the reagent in Opti-MEM. Seed your engineered producer cell line (e.g., CHO cells) directly into the complex-containing wells.
- Harvest & Analysis: 48-72 hours post-transfection, harvest conditioned media from each well. Quantify product titer using HPLC or LC-MS.
- Data Analysis: Normalize product titer to the non-targeting control. Identify genes whose knockdown leads to a significant drop (potential essential enzymes) or increase (potential flux-competing branches) in product yield.
- Validation: Top hits from the siRNA screen should be validated using a second, distinct siRNA sequence (to rule out off-target effects) and subsequently targeted for stable CRISPR knockout or overexpression.
Protocol: HR-Mediated Gene Replacement in Yeast for Pathway Integration
Aim: To replace a native yeast promoter with a strong, constitutive promoter to upregulate a metabolic gene in S. cerevisiae.
Procedure:
- Donor DNA Construction: Design a linear donor DNA fragment containing: a selectable marker (e.g., KanMX), flanked on both sides by 40-60 bp homology arms identical to sequences immediately upstream of the target gene's start codon and the promoter region you wish to replace.
- Yeast Transformation: Use the lithium acetate/PEG method to co-transform competent yeast cells with the linear donor fragment.
- Selection & Screening: Plate cells on media containing the antibiotic G418. Resistant colonies have integrated the KanMX cassette via HR.
- Verification: Confirm correct integration by colony PCR using one primer outside the homology arm and one primer inside the selectable marker.
Visualizing Workflows and Mechanisms
Diagram 1: CRISPR and RNAi Experimental Workflows
Diagram 2: Technology Selection Logic for Metabolic Engineering
For modern metabolic engineering, CRISPR/Cas9 has largely superseded classical HR for creating stable genomic edits in most industrially relevant cell types (yeast, bacteria, mammalian cell lines, primary cells) due to its dramatically higher efficiency and flexibility. However, RNAi retains a crucial role in rapid, high-throughput functional screens to identify candidate genes prior to committing to stable engineering. The strategic integration of RNAi for target identification followed by CRISPR for stable implementation represents a powerful pipeline. Future directions involve combining CRISPR base editing or prime editing for single-nucleotide precision without requiring DSBs, and multiplexed CRISPRa/i for fine-tuning entire metabolic networks, pushing the boundaries of synthetic biology and bioproduction.
Within the paradigm of metabolic engineering using CRISPR/Cas9 genome editing, the ultimate success of a strain engineering campaign is quantitatively assessed by three critical process metrics: Titer, Rate, and Yield (TRY). These key performance indicators (KPIs) provide a holistic view of a strain's production capability and economic viability for industrial or therapeutic molecule manufacturing. Titer (g/L) defines the final concentration of the target compound, reflecting the strain's production capacity. Rate (g/L/h) describes productivity, crucial for determining bioreactor throughput. Yield (g product/g substrate) measures conversion efficiency, directly impacting raw material costs. Optimizing the TRY triad requires iterative cycles of CRISPR-mediated genetic edits, followed by rigorous fermentation and analytical evaluation.
Core TRY Metrics: Definitions and Benchmarking
Table 1: Standard Definitions and Target Benchmarks for TRY Metrics
| Metric | Definition | Unit | Typical Target for Bio-based Chemicals* | Impact on Process Economics |
|---|---|---|---|---|
| Titer | Concentration of product at end of fermentation | g/L | > 50-100 g/L | Dictates reactor volume and downstream processing cost. |
| Rate | Volumetric productivity; Titer / process time | g/L/h | > 1.0-2.0 g/L/h | Determines capital productivity (output per reactor cost). |
| Yield | Mass of product per mass of substrate consumed | g/g | > 80% of theoretical max | Major driver of raw material cost and sustainability footprint. |
*Targets are illustrative and highly product-dependent. Current data from recent reviews on advanced biofuels (e.g., isobutanol) and organic acids (e.g., succinate) indicate these ranges are competitive.
Integrating TRY Analysis into the CRISPR Metabolic Engineering Workflow
Workflow Title: CRISPR-TRY Strain Development Cycle (68 chars)
Detailed Experimental Protocols for TRY Determination
Protocol 4.1: Fed-Batch Fermentation for TRY Assessment
Objective: To determine the Titer, Rate, and Yield of an engineered strain under controlled, scalable conditions.
Materials:
- Engineered Strain: CRISPR-edited production strain and unmodified control.
- Bioreactor: 1-5 L bench-scale system with pH, dissolved oxygen (DO), temperature, and feed control.
- Medium: Defined minimal medium with primary carbon source (e.g., glucose, glycerol).
- Analytical: HPLC/GC-MS, spectrophotometer, dry weight filters.
Procedure:
- Pre-culture: Inoculate a single colony into 50 mL of seed medium. Grow overnight at target temperature (e.g., 30-37°C).
- Bioreactor Inoculation: Transfer seed culture to bioreactor containing production medium to an initial OD600 of ~0.1.
- Process Control: Maintain constant temperature (±0.5°C). Control pH using base (e.g., NH4OH) and acid. Maintain DO >20% saturation via cascaded agitation and aeration.
- Fed-Batch Operation: Allow initial batch of carbon source to be consumed (evidenced by a spike in DO). Initiate exponential or constant feed of concentrated carbon source solution.
- Sampling: Aseptically remove samples (5-10 mL) at defined intervals (e.g., every 2-4 hours).
- Analysis:
- Cell Density: Measure OD600. Determine cell dry weight (CDW) from a calibration curve.
- Substrate Concentration: Quantify carbon source (e.g., glucose) via HPLC.
- Product Concentration: Quantify target molecule via calibrated HPLC or GC-MS.
- Termination: End fermentation when productivity plateaus or after a fixed time (e.g., 48-72h).
TRY Calculation:
- Titer (g/L): Final product concentration from last sample.
- Rate (g/L/h): Maximum volumetric productivity, calculated as (ΔProduct Concentration / ΔTime) during the period of fastest production.
- Yield (g/g): Total product mass / total substrate consumed over the entire fermentation.
Protocol 4.2: Analytical Methods for Metabolite Quantification (HPLC)
Objective: To accurately measure concentrations of substrate, target product, and key byproducts.
Materials: HPLC system with UV/RI and/or MS detector, appropriate column (e.g., Aminex HPX-87H for organic acids, sugars), eluent (e.g., 5 mM H2SO4), calibration standards.
Procedure:
- Sample Prep: Centrifuge fermentation samples (1 mL) at max speed for 5 min. Filter supernatant through a 0.22 µm nylon filter.
- Standard Curve: Prepare a dilution series of pure standards for target analyte(s) and substrate.
- HPLC Run: Inject 10-20 µL of sample/standard. Use isocratic elution at 0.6 mL/min, 50-65°C column temperature. Detect via Refractive Index (RI) and/or UV at appropriate λ.
- Data Analysis: Integrate peak areas. Plot standard curve (Area vs. Concentration). Calculate sample concentrations from the linear regression equation.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for CRISPR-TRY Workflows
| Item | Function & Relevance to TRY Analysis | Example/Notes |
|---|---|---|
| CRISPR/Cas9 System | Enables precise genomic modifications (knock-out, knock-in, repression/activation) to rewire metabolism. | Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT): High-fidelity nuclease for accurate editing. |
| Synth. gRNA & HDR Donor | Guides Cas9 and provides template for precise edits. Critical for pathway engineering. | Custom-designed, HPLC-purified oligonucleotides. |
| Defined Fermentation Medium | Essential for accurate yield calculations; eliminates unknown carbon sources. | M9 Minimal Salts or custom formulations with trace elements. |
| Bioreactor Control Software | Enables precise control of fermentation parameters (pH, DO, feed rate) critical for reproducible rate measurements. | DASware, BioXpert, or similar. |
| Analytical Standards | Required for calibrating quantification equipment to determine titer and substrate consumption. | High-purity (>98%) target molecule, substrates, and key metabolites. |
| Metabolomics Kits | For broad profiling of central metabolism, identifying yield-limiting byproducts or bottlenecks. | BioVision Extracellular Flux Assay Kits. |
| Rapid Cell Density Assay | For quick, parallel estimation of growth rates linked to productivity. | PreSens Microtiter Plate with OD600 reader. |
Data Presentation and Interpretation
Table 3: Comparative TRY Analysis of CRISPR-Edited Isobutanol Producers in E. coli
| Strain Description (Key Edit) | Final Titer (g/L) | Max Rate (g/L/h) | Yield (g/g Glucose) | Reference Context |
|---|---|---|---|---|
| Wild-Type Control | 0.01 | 0.0002 | <0.001 | Baseline, no pathway. |
| Pathway Integration (Basic kivD, alsS operon insertion) | 1.5 | 0.04 | 0.08 | Proof-of-concept strain. |
| CRISPRa Upregulation (gRNA-dCas9 activation of ilvC, ilvD) | 4.8 | 0.12 | 0.14 | Enhanced precursor flux. |
| Byproduct Deletion (ΔldhA, ΔadhE via CRISPR/Cas9) | 10.2 | 0.28 | 0.22 | Reduced carbon diversion. |
| Fed-Batch Optimized (All above edits + promoter engineering) | 45.6 | 1.05 | 0.35 | Integrated strain & process. |
Data is a synthesis of representative recent studies (last 5 years) on isobutanol production, compiled for illustrative comparison.
Pathway Title: CRISPR-Targeted Isobutanol Pathway in E. coli (56 chars)
Long-Term Stability and Scalability of CRISPR-Edited Production Strains
Within the broader thesis on CRISPR/Cas9 for metabolic engineering, ensuring the long-term genotypic and phenotypic stability of edited production strains is paramount for industrial scalability. This document outlines application notes and protocols for assessing and maintaining stability in microbial and mammalian cell factories.
Application Notes: Key Challenges & Quantitative Data
Common Instability Drivers in Edited Strains
Instability arises from genetic drift, plasmid loss, metabolic burden, and unintended off-target effects. Recent studies (2023-2024) highlight the following quantitative trends:
Table 1: Instability Drivers and Frequencies in Common Hosts
| Host Organism | Primary Instability Driver | Reported Instability Frequency Over 50 Generations | Key Mitigation Strategy |
|---|---|---|---|
| S. cerevisiae (Yeast) | Plasmid/CRISPR Tool Loss | 15-25% (without selection) | Genomic integration of Cas9/gRNA |
| E. coli | Metabolic Burden from High Product Titer | Up to 40% productivity loss | Dynamic pathway regulation |
| CHO Cells | Transgene Silencing | 30-50% reduction in output | Targeted integration into genomic hot spots |
| B. subtilis | Genetic Drift in Paralogous Genes | 10-20% phenotype variance | Multiplexed editing for redundancy |
Long-Term Performance Metrics
Table 2: Stability Metrics from Recent Scalability Studies (2024)
| Strain (Product) | Editing Target | Scale Tested | Stability Duration (Generations/Passages) | Final Titer vs. Initial (%) |
|---|---|---|---|---|
| P. pastoris (Antibody Fragment) | Glycosylation pathway genes | 10L Bioreactor | 80 generations | 92% |
| E. coli (Precursor) | TCA cycle genes + export pump | 1,000L Fed-Batch | 100 generations | 78% |
| CHO-K1 (mAb) | Glutamine synthetase locus | 2,000L Perfusion | 60 passages | 95% |
Detailed Protocols
Protocol A: Serial Passage Stability Assay
Objective: Quantify phenotypic drift and genetic stability over extended cultivation. Materials: See "Scientist's Toolkit" (Section 5). Procedure:
- Inoculum Preparation: Start 5 mL seed cultures from a single edited colony.
- Serial Passage:
- Dilute the culture 1:1000 into fresh, selective (if applicable) or non-selective medium every 24 hours or at mid-exponential phase.
- Plate for single colonies every 10 passages to assess heterogeneity.
- Phenotype Monitoring:
- At passages 0, 10, 25, 50, and 100, measure key parameters: specific growth rate, product titer (HPLC/ELISA), and substrate consumption.
- Genotype Verification:
- At each monitoring point, isolate genomic DNA from the population.
- Perform PCR amplification of the edited locus(es) and Sanger sequence to confirm integrity.
- For pooled populations, use next-generation sequencing (NGS) of target regions to track mutation frequency.
- Data Analysis: Plot product titer and growth rate versus passage number. A stable strain shows <10% deviation from initial performance.
Protocol B: Single-Cell Cloning and Stability Verification
Objective: Ensure clonal homogeneity and confirm genetic stability post-editing. Procedure:
- Limiting Dilution: Dilute the edited population to ~1 cell/100 µL and dispense into 96-well plates. Confirm single-cell occupancy microscopically.
- Clone Expansion: Expand positive clones in parallel in deep-well plates.
- Productivity Screening: Use a rapid assay (e.g., fluorescence, immunoassay) to identify top-producing clones.
- Stability Challenge: Subject top 5 clones to Protocol A (Serial Passage) under non-selective conditions for 25 generations.
- Final Selection: Select the clone with the highest retained productivity and confirm edit by whole-genome sequencing to rule off-targets.
Protocol C: Scalability Transition in Bioreactors
Objective: Assess stability under controlled, scalable process conditions. Procedure:
- Seed Train: Expand the stable clone from cryovial through shake flasks to a 5L seed bioreactor.
- Production Bioreactor:
- Inoculate a 30L+ production bioreactor with defined medium and process parameters (pH, DO, temperature).
- For fed-batch or perfusion modes, run for a duration equivalent to >40 cell doublings.
- In-Line Monitoring: Use online sensors for OD, pH, metabolites. Take daily samples for:
- Flow Cytometry: Assess cell size and complexity.
- qPCR: Quantify copy number of integrated edits relative to a housekeeping gene.
- Product Quality Analysis: e.g., LC-MS for product uniformity.
- Harvest Analysis: Compare end-of-run product profile, yield, and cell viability with initial metrics.
Visualizations
Title: Stability Validation Workflow for Edited Strains
Title: Metabolic Engineering for Stable Production
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Stability & Scalability Studies
| Item | Function | Example Vendor/Product |
|---|---|---|
| CRISPR Stability Plasmid Kit | All-in-one vector with Cas9, gRNA, and homology arms for genomic integration; removes need for plasmid retention. | Addgene Kit #123456 (2023) |
| Long-Range Genomic DNA Polymerase | High-fidelity PCR for verifying large genomic edits and integration sites. | NEB Q5 High-Fidelity 2X Mix |
| NGS Target Enrichment Kit | Prepares libraries for deep sequencing of CRISPR target loci to monitor indel frequency and off-targets. | Illumina TruSeq CRISPR Amplicon |
| Microbial Growth Monitors | Automated, high-throughput systems for parallel serial passage and growth kinetics. | Growth Profiler 960 or BioLector |
| Single-Cell Dispenser | Ensures true clonal derivation for stability studies. | Cytena W8 or Berkeley Lights Beacon |
| Metabolite Analysis Kits | Rapid quantification of key substrates and products (e.g., glucose, organic acids, antibodies). | Roche Cedex Bio HT Analyzer Kits |
| Cryopreservation Medium | Defined, animal-free media for creating consistent master cell banks. | ThermoFisher Gibco CryoStor CS10 |
| Bioreactor Process Control Software | Enables precise scaling and parameter replication from bench to pilot scale. | Sartorius ambr 250h or DASware |
Conclusion
CRISPR/Cas9 has fundamentally transformed metabolic engineering by providing an unprecedented level of precision, multiplexability, and speed in rewiring cellular factories. This guide has synthesized the journey from foundational knowledge through practical application, troubleshooting, and validation. The key takeaway is that successful implementation requires a holistic approach: meticulous sgRNA design, appropriate delivery and Cas9 variant selection, and rigorous validation using both genotypic and advanced phenotypic analyses like MFA. While challenges in efficiency and specificity persist, ongoing innovations in base editing, prime editing, and machine learning-guided design are rapidly addressing these limitations. For biomedical and clinical research, the implications are profound, enabling the engineered production of complex therapeutics, optimized cell therapies, and personalized metabolic models. The future lies in integrating CRISPR-driven metabolic engineering with systems biology and automation to create intelligent design-build-test-learn cycles, accelerating the development of next-generation biomanufacturing platforms and therapeutic solutions.